29 October 2020: Articles 
Different Phenotypes of Anderson-Fabry Disease Identified with Cardiac Magnetic Resonance Imaging in a Family with the Same Late-Onset Mutation
Unusual clinical course, Challenging differential diagnosis, Rare disease
Diego A. Ávila-Sánchez1ABDEF*, Esther Cambronero-Cortinas1ADE, Manuel Barreiro-Pérez1BDE, Juan L. Rodríguez-Hernández1BF, Brais Díaz-Fernández1CF, Pedro L. Sánchez1ADOI: 10.12659/AJCR.925631
Am J Case Rep 2020; 21:e925631






Abstract
BACKGROUND: Cardiac magnetic resonance imaging (CMR) is the only noninvasive test capable of differentiating between hypertrophic cardiomyopathy (HCM) and late-onset Anderson-Fabry disease (AFD). The purpose of this report is to show how CMR led to diagnosis of AFD in 3 family members, 1 of whom previously was misdiagnosed with HCM, and how late-onset AFD can present with different cardiac phenotypes, even in a family with the same pathogenic mutation.
CASE REPORT: A 60-year-old man was referred because of evidence of left ventricular hypertrophy (LVH) on an electrocardiogram (ECG) that was performed to screen for cardiomyopathy. One of his siblings previously had been diagnosed with HCM and atrial fibrillation. The patient’s ECG and echocardiographic findings were suspicious for HCM. CMR showed severe symmetrical LVH but tissue characterization sequences were highly suggestive of AFD cardiomyopathy. Enzymatic and genetic testing confirmed the diagnosis of late-onset AFD (presence of the GLA p.F113.L mutation). The brother of the index patient then was re-evaluated and also diagnosed with late-onset AFD. He was found to have the same pathogenic mutation but with a presentation of asymmetrical septal LVH. The daughter of the index patient was positive for the same mutation but did not have LVH.
CONCLUSIONS: The fact that patients with late-onset AFD can present with different LVH and fibrosis patterns, even in the presence of the same pathogenic mutation, underscores the importance of including AFD in the differential diagnosis of HCM. CMR is fundamental for differentiating between those 2 entities and defining the pathological phase of AFD. A correct diagnosis can have a substantial impact on patient management, and more so on thier families.
Keywords: Cardiomyopathies, Fabry Disease, Hypertrophy, Left Ventricular, Magnetic Resonance Imaging, Cardiomyopathy, Hypertrophic, Mutation, Phenotype
Background
The etiology of concentric left ventricular hypertrophy (LVH) is heterogeneous, and its causes include chronic pressure overload (arterial hypertension, aortic stenosis), hypertrophic cardiomyopathy (HCM), and infiltrative cardiomyopathies such as amyloidosis and Anderson-Fabry disease (AFD). AFD has been reported in 6% of men [1] and 12% of women [2] previously diagnosed with HCM. In fact, late-onset AFD is indistinguishable from HCM based on clinical symptoms or findings from an electrocardiogram (ECG) or echocardiogram [3]. In a patient with LVH, cardiac magnetic resonance imaging (CMR) is the only noninvasive test capable of differentiating between HCM and late-onset AFD. The purpose of this report is to show how CMR led to diagnosis of AFD in 3 family members, 1 of whom previously was misdiagnosed with HCM, and how late-onset AFD can present with different cardiac phenotypes on CMR, even in family members who have the same AFD pathogenic mutation.
Case Report
A 60-year-old man with no known medical history was referred to our cardiology clinic because findings from a previous ECG met the criteria for LVH (Figure 1) and screening for cardiomyopathy was suspicious for HCM. He had a family history of cardiovascular disease, and 1 sibling had been diagnosed with HCM and paroxysmal atrial fibrillation. The patient’s only complaint was episodes of palpitation that lasted a few minutes. A 24-hour Holter tape revealed sinus rhythm (mean heart rate 78 bpm) and low-density ventricular and supraventricular premature beats with no ventricular tachycardia. Transthoracic echo-cardiography (TTE) showed severe symmetrical concentric LVH (18-mm thickness at the interventricular septum) and a higher than normal left ventricular ejection fraction (LVEF; 74%), with no regional wall motion abnormalities. There was no systolic anterior motion of the mitral valve or left ventricular (LV) out-flow tract obstruction at rest. An LV myocardial deformation study showed decreased global longitudinal strain (GLS) (–15%) (Figure 2). Because of the patient’s family history and his ECG and TTE findings, there was significant suspicion for symmetrical non-obstructive HCM. CMR was performed, which confirmed the echocardiographic findings (Figure 3, Videos 1–3). Late gadolinium enhancement (LGE) sequences documented mid-wall fibrosis in the basal to mid-lateral segments (Figures 4, 5). Furthermore, the myocardial T1 mapping sequence showed a low native T1 value of 830 milliseconds at the mid-septum at 1.5 Tesla (the normal range in our center is between 959 milliseconds and 1031 milliseconds), with normal extracellular volume (ECV) of 22% (the normal range in our center is <28%). Myocardial T2 star values were normal (39 milliseconds), excluding iron overload. All of these findings were highly suggestive of AFD cardiomyopathy, for which an enzymatic and genetic study was carried out, revealing a low alpha galactosidase (α-Gal A) protein concentration (<2.8 ng/mL, the lowest quantification limit detected; normal range >15.3 ng/mL) and increased lyso-GB3 levels. Moreover, the genetic study was positive for heterozygosis mutation NM_000169.2: c.337T>C; p.Phe113Leu in the
The patient’s renal function was normal and he did not have any other extracardiac manifestations. Therefore, Type 2 AFD (late-onset variant) was diagnosed and screening of other relatives was indicated. The index patient had 4 brothers and 1 daughter. His 58-year-old brother previously had been diagnosed with HCM, based on his clinical history and ECG and echocardiographic findings. His CMR showed asymmetrical septal LVH (anteroseptal wall thickness 16 mm
Based on these findings, the 3 family members were diagnosed with late-onset AFD (the brother’s HCM diagnosis was disregarded) and referred for evaluation for specific AFD therapy and familial screening. The 2 men were started on enzyme replacement therapy (ERT), angiotensin-converting enzyme inhibitors (ACEIs), and beta blockers. For the daughter, we chose to continue with a conservative strategy, with close regular follow-up, defined as an annual ECG and echocardiogram and 5-year interval CMR imaging. If she had cardiac symptoms or new abnormalities on ECG or echocardiography, we would perform CMR imaging sooner.
Of the index patient’s other 3 brothers, 1 is healthy and 2 were diagnosed with LVH at another center. Diagnostic studies for them are still pending.
Discussion
AFD is a rare, X-linked lysosomal storage disorder caused by deficiency in the enzyme α-galactosidase A [5], which leads to progressive sphingolipid accumulation that can affect multiple organs and systems, including the heart, kidneys, eyes, skin, and brain. The disease has 2 main phenotypes: the classic form (Type 1; no α-galactosidase A activity), a multisystemic disease that presents in childhood or adolescence and is more frequent in males; and the late-onset variant (Type 2; some residual α-galactosidase A activity), commonly found in women or in some men who have specific mutations [6,7]. In the later variant, heterozygote patients have a lower amount of functional α-Gal A, albeit with some residual activity, which causes late manifestation of the disease in the fifth to seventh decades of life, typically affecting a single organ, often the heart or kidney [6,7].
Type 2 AFD is 20 times more prevalent than the classic pheno-type (approximately 1: 1000
The current guideline from the American College of Cardiology/ American Heart Association for diagnosis and treatment of HCM [10] recommends CMR imaging (Class I, Level B) for patients with suspected HCM if echocardiography is inconclusive or in those with known HCM if the treatment decision (septal myectomy or alcohol septal ablation) may be influenced by additional information. However, the recommendation is weak (Class IIb, Level C) for the use of CMR imaging in patients with LVH and the suspicion of an alternative diagnosis (like AFD). The current European Society of Cardiology HCM guideline [11] does not even make a recommendation for the latter scenario. We strongly believe that CMR imaging should have a higher level of recommendation in patients with LVH when there is a suspicion of infiltrative cardiomyopathies such as AFD or cardiac amyloidosis.
Regarding LGE with CMR, the presence and location of focal fibrosis or any infiltration data is useful in the assessment of LVH and can suggest a specific etiology. For example, LGE is present in 60% to 70% of individuals with HCM, and the most common pattern is patchy and intramyocardial fibrosis in the septum or most hypertrophied segments [12]. Conversely, in AFD, the most described pattern is intramyocardial fibrosis located mainly at the basal to mid-lateral wall in 50% to 80% of patients, but this finding is not AFD-specific and can be present with other conditions [3,13]. Moreover, in both AFD and HCM, LGE reflex inflammation and fibrosis, but it may be absent in the early phases and diffuse and more extensive in advanced stages, complicating the differential diagnosis.
The best CMR tool for identifying AFD cardiomyopathy is mapping. Native T1 mapping values are decreased in AFD, reflecting accumulation of glycosphingolipids in the myocardium. It has been shown that a lower T1 value in the context of LVH has excellent sensitivity and specificity for identification of patients with AFD and can completely differentiate this disease from other etiologies in which values are normal or elevated, such as HCM, cardiac amyloidosis, hypertensive heart disease, and aortic stenosis [14]. Iron overload is the only other condition in which native T1 is reduced, but it does not produce LVH and can be correctly assessed with T2* mapping. With T1 mapping, it is also possible to calculate the extracellular volume (ECV). In AFD, the ECV is normal (sphingolipid accumulation is intracellular) and in HCM, it is usually normal or high-normal (in relation to myocardial fibrosis).
Besides the focal inferolateral fibrosis and low native T1 values typically described in AFD (and present in our index patient and his brother), a recent study described 3 phases of phenotype evolution in AFD cardiomyopathy [15]: an early phase (accumulation phase), which presents with normal or low native T1, and without LVH or LGE; an intermediate phase (inflammatory and hypertrophic phase) characterized by low native T1, some degrees of LVH, and focal mid-wall inferolateral fibrosis; and an advance phase (extensive fibrosis and LV impairment) with pseudonormalization of native T1 value secondary to diffuse LGE (fibrosis elevates T1 values), persistent LVH (with some areas of myocardial thinning), and systolic impairment. It is important to consider that in the intermediate phase, the focal fibrosis can lead to what appears to be a normal native T1 value in the basal to mid-inferolateral wall, but low values elsewhere.
In the present report, we describe 3 family members with the same AFD pathogenic mutation (heterozygote, p.F113L mutation), but with different cardiac phenotypes on CMR. The 28-year-old woman is in the accumulation phase (normal T1, negative LVH), whereas her 58- and 60-year-old brothers are in the hypertrophic and inflammatory phases of the disease (low T1, positive LVH, positive LGE), respectively. The differences between the woman and the men are in accord with previous studies on late-onset AFD, which reported that clinical phenotype is critically influenced by gender and age. For example, LVH development occurs much later in women (complete penetrance by 60 to 69 years in them compared with 40 to 49 years in men), and when present, it is more severe in men [15,18]. Besides that, the reduction in native T1 with age is steeper in men than in women, which suggests that storage is faster in men. These sex-based differences in disease course are mainly caused by 2 factors: first, the women have a second functional copy of the alpha galactosidase gene; and second, there is a sex dimorphism related to the male myocyte response to the insult, probably related to different expression of androgen and estrogen receptors and differences in the renin-angiotensin system, nitric oxide activity, and norepinephrine release [16].
Although in our report, both brothers are in the same phase of the disease and have an identical AFD pathogenic mutation, they have different cardiac phenotypes. The index patient has severe symmetrical LVH and positive LGE and his brother has asymmetrical septal LVH without LGE (Figure 7, Table 1). Regarding these differences, a report on a previous cohort [13] described different LVH patterns in patients with the classical form of the disease, with a 44% prevalence of concentric symmetrical LVH and 8% prevalence of asymmetric septal LVH. The latter form was associated with a thicker wall and higher left ventricle mass index (LVMI), whereas in our study, the asymmetric septal LVH pattern was seen in the individual with a thinner wall and lower LVMI (Table 1). The LGE prevalence in late-onset AFD reportedly is between 40% and 70% [13,17,18], and higher in patients who are older and have higher LVH. That is in keeping with the findings from the present study, because positive LGE was found only in the patient with severe LVH. The different cardiac features between brothers evidence that late-onset AFD phenotype is influenced not only by the underlying pathogenic mutation, sex or age, but also by individual polygenic heritage and environmental risk factors, which produces a complex pathophysiologic cascade pathway that finally determines the disease expression [19].
Recently, Azevedo et al. [17,18] described the clinical profile and natural history of late-onset AFD caused by the p.F113L mutation. They studied 203 consecutive patients with AFD who had that specific mutation and demonstrated that cardiac manifestations have the highest prognostic impact. The first cardiac manifestations are LVH and LGE, which begin in men older than age 30 years and are followed by heart failure (HF), non-sustained ventricular tachycardia (VT), and cardiac conduction disorders such as bifascicular block and complete atrioventricular (AV) block in patients who are older than age 50 years [18]. In women, cardiac manifestations start 2 decades later and also increase with age. LVH mass on MRI is a predictor of LGE (P<0.001), but interestingly, LGE can be present even in patients who do not have LVH (LGE prevalence of 52% in patients with LVH
Treatment options for AFD include ERT, which has been the mainstay therapy, and oral chaperone therapy [21], which is available for amenable mutations. When to start therapy for late-onset AFD in men and women is unclear. In general, ERT is indicated when there is laboratory, histological or imaging evidence of injury to the heart or when symptoms of cardiac disease are present [22], but some studies suggest that ERT should be considered in the preclinical phase before cardiac manifestations occur [9]. There is evidence that it is best to administer ERT before development of myocardial fibrosis to maintain long-term myocardial morphology and function [23]. It is important to consider adjunctive therapy with ACEI, angiotensin receptor blockers, and beta blockers and to avoid amiodarone. Even though our index patient was diagnosed when he had severe LVH, focal inferolateral fibrosis, and LV function impairment (abnormal myocardial strain with GLS –15%), and the clinical benefits of ERT probably are limited in this scenario, we decided to prescribe it because it is safe and there is no strong evidence contraindicating its use in this context. ERT also was prescribed to the index patient´s brother. In his case, the benefit was greater because he did not have myocardial fibrosis, and in that scenario, the treatment can reduce LVH and improve myocardial function and exercise capacity [23]. Given the natural history of the mutation in the index patient’s daughter, meaning that cardiac manifestations usually arise in women who are older than age 40 to 50 years, we decided on expectant management with close follow-up in our Inherited Cardiomyopathies Clinic and periodic cardiac imaging. The goal for her is to identify early cardiac manifestations of the disease. More clinical trials are needed of results with ERT for late-onset AFD cardiomyopathy. Timing of initiation of therapy should be individualized to each patient to avoid target organ damage and improve clinical results.
Conclusions
The fact that late-onset AFD can present with different LVH and fibrosis patterns, even in a family with the same pathogenic mutation, underscores the importance of including the disease in the differential diagnosis of HCM. CMR with LGE and mapping sequences is fundamental to distinguish between those entities. CMR also can precisely define the pathological phase of AFD, which provides prognostic information and can be used to guide specific therapy. The correct diagnosis may have a substantial impact on management of patients, and more so on management of their families.
Figures
References:
1.. Sachdev B, Takenaka T, Teraguchi H, Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy: Circulation, 2002; 105; 1407-11
2.. Chimenti C, Pieroni M, Morgante E, Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy: Circulation, 2004; 110; 1047-53
3.. De Cobelli F, Esposito A, Belloni E, Delayed-enhanced cardiac MRI for differentiation of Fabry’s disease from symmetric hypertrophic cardiomyopathy: AmJ Roenrgenol, 2009; 192; 97-102
4.. Oliveira JP, Nowak A, Barbey F, Fabry disease caused by the GLA p.Phe113Leu (p.F113L) variant: Natural history in males: Eur J Med Genet, 2020; 63(2); 103703
5.. Germain DP, Fabry disease, orphanet: J Rare Dis, 2010(5); 5-30
6.. von Scheidt W, Eng CM, Fitzmaurice TF, An atypical variant of Fabry’s disease with manifestations confined to the myocardium: N Engl J Med, 1991; 324; 395-99
7.. Nakao S, Takenaka T, Maeda M, An atypical variant of Fabry’s disease in men with left ventricle hypertrophy: N Engl J Med, 1995; 333; 288-93
8.. Bokhari SR, Hariz A, Fabry disease: StatPearls Publishing, 2019 www.ncbi.nlm.nih.gov/books/NBK435996/
9.. Hsu TR, Hung SC, Chang FP, Late onset Fabry disease, cardiac damage progress in silence. Experience with a highly prevalent mutation: J Am Coll Cardiol, 2016; 68(3); 2554-63
10.. Gersh BJ, Maron BJ, Bonow RO, 2011 ACCF/AHA Guideline for the diagnosis and treatment of hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines: Circulation, 2011; 124; 783-831
11.. Elliot PM, Anastasakis A, Borger MA, 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC): Eur Heart J, 2104; 35(39); 2733-79
12.. Conte MR, Bongioanni S, Chiribiri A, Late gadolinium enhancement on cardiac magnetic resonance and phenotypic expression in hypertrophic cardiomyopathy: Am Heart J, 2011; 161; 1073-77
13.. Deva DP, Hanneman K, Li Q, Cardiovascular magnetic resonance demonstration of the spectrum of morphological phenotypes and patterns of myocardial scarring in Anderson-Fabry disease: J Cardiovasc Magn Reson, 2016; 18; 14
14.. Sado DM, White SK, Piechnik SK, Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping: Circ Cardiovasc Imaging, 2013; 6; 392-98
15.. Nordin S, Kozor R, Medina-Menacho K, Proposed stages of myocar-dial phenotype development in Fabry disease: JACC Cardiovasc Imaging, 2019; 12(8); 1673-83
16.. Marsh JD, Lehmann MH, Ritchie RH, Androgen receptors mediate hypertrophy in cardiac myocytes: Circulation, 1998; 98; 256-61
17.. Azevedo O, Gal A, Faria R, Founder effect of Fabry disease due to p.F113L mutation: Clinical profile of a late-onset phenotype: Mol Genet Metab, 2020; 129(2); 150-60
18.. Azevedo O, Gago M, Miltenberger-Miltenyi G, Natural history of the late-onset phenotype of Fabry disease due to the p.F113L mutation: Mol Genet Metab Rep, 2020; 22; 1-11
19.. Oliveira JP, Ferreira S, Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype-phenotype correlations: Appl Clin Genet, 2019; 12; 35-50
20.. Teraguchu H, Takenake T, Yoshida A, End-stage cardiac manifestations and autopsy findings in patient with cardiac Fabry disease: J Cardiol, 2004; 43; 98-99
21.. Germain DO, Hughes DA, Nicholls K, Treatment of Fabry’s disease with the pharmacologic chaperone migalastat: N Engl J Med, 2016; 375; 545-55
22.. Ortiz A, Germain DP, Desnick RJ, Fabry disease revisited: Management and treatment recommendations for adult patients: Mol Genet Metabol, 2018; 123; 416-26
23.. Wiermann F, Niemann M, Breunig F, Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: Evidence for a better outcome with early treatment: Circulation, 2009; 119; 524-29
Figures
Tables
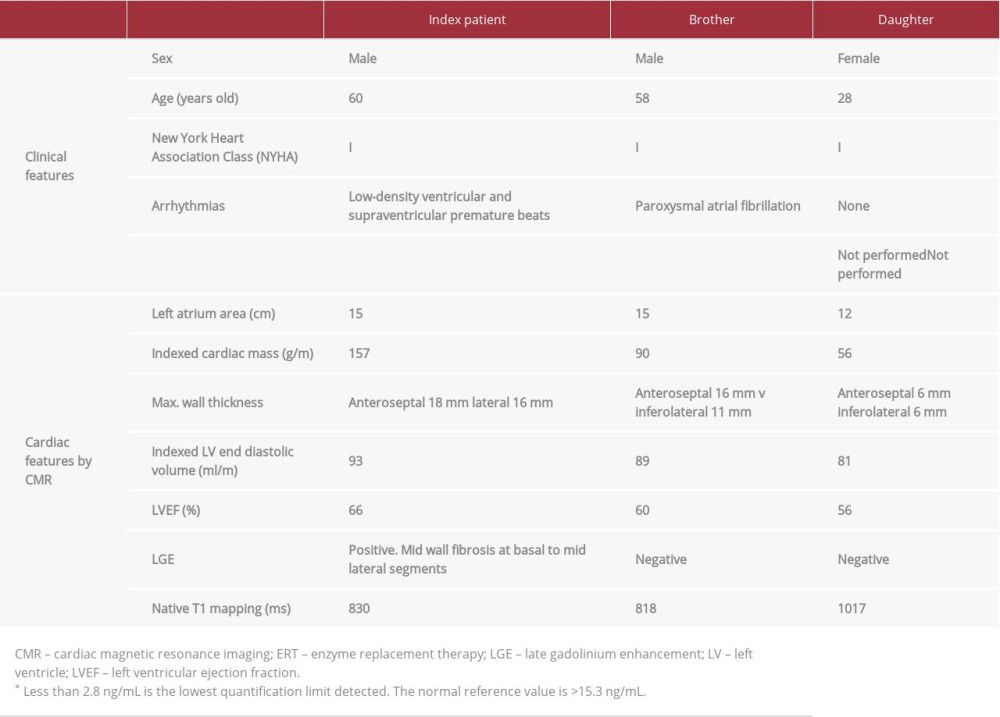 Table 1.. The main clinical and cardiac features by cardiac magnetic resonance imaging in 3 family members with late-onset Anderson-Fabry disease and the same p.F113L pathogenic mutation.Table 1.. The main clinical and cardiac features by cardiac magnetic resonance imaging in 3 family members with late-onset Anderson-Fabry disease and the same p.F113L pathogenic mutation.
Table 1.. The main clinical and cardiac features by cardiac magnetic resonance imaging in 3 family members with late-onset Anderson-Fabry disease and the same p.F113L pathogenic mutation.Table 1.. The main clinical and cardiac features by cardiac magnetic resonance imaging in 3 family members with late-onset Anderson-Fabry disease and the same p.F113L pathogenic mutation. In Press
14 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942966
05 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942032
06 Mar 2024 : Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.942937
12 Mar 2024 : Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.943244
Most Viewed Current Articles
07 Mar 2024 : Case report 
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250