17 August 2021: Articles 
Drug-Associated Eosinophilic Fasciitis: A Case of Eosinophilic Fasciitis Secondary to Cemiplimab Therapy
Challenging differential diagnosis, Unusual or unexpected effect of treatment, Unexpected drug reaction, Rare disease, Clinical situation which can not be reproduced for ethical reasons
Sri Harsha Boppana1A*, Nageswara Rao Dulla2EF, Bryce D. Beutler3EF, Nageshwara Gullapalli1ADEFG, Ratinder Kaur14DEDOI: 10.12659/AJCR.932888
Am J Case Rep 2021; 22:e932888






Abstract
BACKGROUND: Eosinophilic fasciitis, also known as Shulman syndrome, is a rare inflammatory condition characterized by diffuse erythema and progressive collagenous thickening of the subcutaneous fascia. The underlying cause remains to be definitively established; however, several drugs have been linked to this uncommon clinical entity. We present a rare case of eosinophilic fasciitis secondary to immune checkpoint inhibitor therapy.
CASE REPORT: A 72-year-old woman with metastatic cutaneous squamous cell carcinoma presented to the rheumatology clinic for evaluation of joint pain that developed 3 weeks after beginning treatment with cemiplimab. The correlation of clinical history and physical examination was most consistent with osteoarthritis. Symptoms improved after a short course of low-dose prednisone. The patient continued cemiplimab therapy for approximately 1 year and was subsequently transitioned to carboplatin and radiation therapy. However, relapse occurred shortly thereafter, and cemiplimab was restarted. Two weeks later, the patient developed severe joint pain, morning stiffness, and extensive cutaneous discoloration and induration. A skin biopsy was performed. Microscopic examination of a tissue sample showed a mononuclear infiltrate with plasma cells and eosinophils. A diagnosis of eosinophilic fasciitis was established. Cemiplimab was held and the patient was treated with hydroxychloroquine, prednisone, and sulfasalazine. Symptoms improved within 1 week.
CONCLUSIONS: Eosinophilic fasciitis is a rare but important adverse effect of immune checkpoint inhibitors. Individuals receiving immunotherapy should be monitored closely for symptoms of eosinophilic fasciitis, as prompt treatment is essential to prevent long-term complications.
Keywords: Drug Eruptions, eosinophilic fasciitis, Neoplasms, Squamous Cell, Antibodies, Monoclonal, Humanized, Carcinoma, Squamous Cell, Eosinophilia, Fasciitis, Pharmaceutical Preparations, Skin Neoplasms
Background
Eosinophilic fasciitis (EF), previously known as Shulman syndrome, is a rare inflammatory condition characterized by erythema and edema of the trunk and limbs, followed by diffuse collagenous thickening of the subcutaneous fascia [1]. Hypergammaglobulinemia and eosinophilia are common. Interestingly, while hypergammaglobulinemia typically persists throughout the course of EF, eosinophilia may decrease and even resolve as the disease progresses [2]. Young adult males are most commonly affected. However, EF has also been reported in children and the elderly [3].
The pathogenesis of EF remains to be definitively established, but it has been linked to
The classic clinical feature of EF is “woody induration,” characterized by skin thickening and orange-colored hyperpigmentation involving the trunk and/or extremities. Localized morphea, defined as localized inflammation in the reticular dermis and superficial panniculus, has also been reported. Muscle weakness and rigidity is common. Rarely, synovitis and contractures can occur. EF shares similarities with many other diseases, which often leads to a delayed diagnosis [10]. We present a patient with EF that developed after beginning a second cycle of cemiplimab for metastatic squamous cell carcinoma.
Case Report
A 72-year-old woman with metastatic cutaneous squamous cell carcinoma involving the parotid gland and thoracic vertebral bodies presented to the rheumatology clinic for evaluation of joint pain that developed 3 weeks after beginning treatment with cemiplimab. The patient reported having mild pain and stiffness in both hands, which was most severe in the afternoon and evening and worsened with exertion. She denied any other symptoms, including joint swelling, fatigue, xerostomia, dysphagia, and skin changes. There was no past medical history of auto-immune or hematologic disease. The patient denied recent exercise or rigorous exertion. Physical examination revealed small Herberden nodes affecting the first 2 digits on each hand, but was otherwise unremarkable. Laboratory studies showed elevated titers of anti-nuclear antibody (1: 1280), rheumatoid factor (101 IU/mL), and SSA antibody (64 AU/mL). However, given the patient’s clinical history and physical examination findings, an autoimmune etiology for her joint pain was considered less likely, and a presumptive diagnosis of osteoarthritis was established. The patient was advised to take non-steroidal anti-inflammatory drugs (NSAIDs) as needed and follow-up for further investigation if symptoms worsened. The pain and stiffness was markedly improved after several weeks of treatment with NSAIDs.
The patient continued cemiplimab therapy for approximately 1 year and was subsequently transitioned to carboplatin and radiation therapy. However, relapse occurred shortly thereafter and cemiplimab was restarted. Two weeks later, the patient developed severe joint pain, morning stiffness, skin discoloration, and woody induration extending along her bilateral forearms to the wrist, with sparing of the distal forearms and hands (Figure 1). Depression was seen along the course of the basilic veins, consistent with the Groove sign. The patient reported that she could no longer extend her elbows without severe pain. Physical examination revealed full range of motion of the cervical spine, with normal flexion, extension, and lateral movement. Examination of the lumbosacral spine was also normal. There was normal abduction, flexion, extension, external rotation, and internal rotation of the bilateral hips and shoulders. Bilateral hand grip strength was decreased (3/5 on the left and 4/5 on the right). Laboratory studies, including hemoglobin, white blood cells, platelets, electrolytes, and calcium, were within normal limits; the absolute eosinophil count was 0.28 K/uL (reference range, 0.1–0.5 K/uL); erythrocyte sedimentation rate was 3 mm/h (reference range, 0–30 mm/h); serum creatine phosphokinase level was 35 U/L (reference range, 26–192 U/L); and aldolase was 4.7 U/L (reference range, 1.0–7.5 U/L). A full-thickness skin biopsy sample of an affected region on the patient’s right forearm was obtained. Microscopic examination showed a mononuclear infiltrate with plasma cells and eosinophils (Figures 2, 3). Based on the correlation of clinical, laboratory, and histopathologic findings, a diagnosis of eosinophilic fasciitis was established.
Other differential considerations were considered less likely; the pattern of skin involvement was atypical for systemic sclerosis, and myositis was excluded based on the normal serum creatine phosphokinase and aldolase levels.
Cemiplimab was held and methotrexate treatment was recommended. However, the patient refused, expressing concern that methotrexate would increase her risk of developing a second malignancy. She was therefore treated with hydroxychloroquine 200 mg twice daily, prednisone 40 mg once daily, and sulfasalazine 500 mg twice daily. Her symptoms improved within 1 week and resolved within 1 month. The patient subsequently resumed cemiplimab at a reduced dose, with biweekly monitoring for adverse effects.
Discussion
Immune checkpoints are inhibitory immune pathways that maintain self-tolerance by regulating the immune responses in tissues [11]. Among immune checkpoints, the role of PD-1, its ligand PDL-1, and cytotoxic T-lymphocyte-associated protein 4 in immune regulation is well known. Cemiplimab is a humanized monoclonal antibody that blocks the PD1/PDL1 pathway and is approved for the treatment of squamous cell carcinoma [12,13].
While arthralgia, myalgia, and arthritis are musculoskeletal manifestations of checkpoint inhibitors (CPI), scleroderma or scleroderma-like manifestations are less likely to be seen with the use of these drugs. They are classified under non-musculo-skeletal rheumatic immune-related adverse events [14–16]. EF is an extremely rare condition classified under scleroderma-like disorders. Face and hands are generally spared in EF [17,18]. In our patient, there was the involvement of bilateral upper and lower extremities. Full-thickness biopsy sample analysis from the affected sites showed fibrosis of the subcutaneous connective tissue, cellular infiltration by eosinophils and monocytes, and thickening of the fascia (Figures 2, 3). However, the presence of eosinophils on a microscopic exam is not necessary for diagnosis [19]. Another differential diagnosis for EF is progressive systemic sclerosis, which can be excluded based on clinical features rather than pathological features.
Diagnostic criteria for EF were established by Jinnin et al [19]. Major criteria include (1) symmetric or asymmetric swelling, in-duration and thickening of the skin or subcutaneous tissues and (2) full-thickness wedge biopsy showing fascial thickening with lymphocytes and macrophages, with or without eosinophils. Minor criteria include (1) peripheral eosinophilia; (2) hypergammaglobulinemia; (3) muscle weakness; (4) Groove sign; and (5) magnetic resonance imaging showing T2 hyperintensity of the fascia. Importantly, cutaneous manifestations of EF can be absent in some patients or may not appear until late in the course of the disease. Laboratory studies to assess for eosinophilia and hypergammaglobulinemia and magnetic resonance imaging of the fascia can therefore play an important role in establishing a diagnosis of EF among patients without characteristic skin findings.
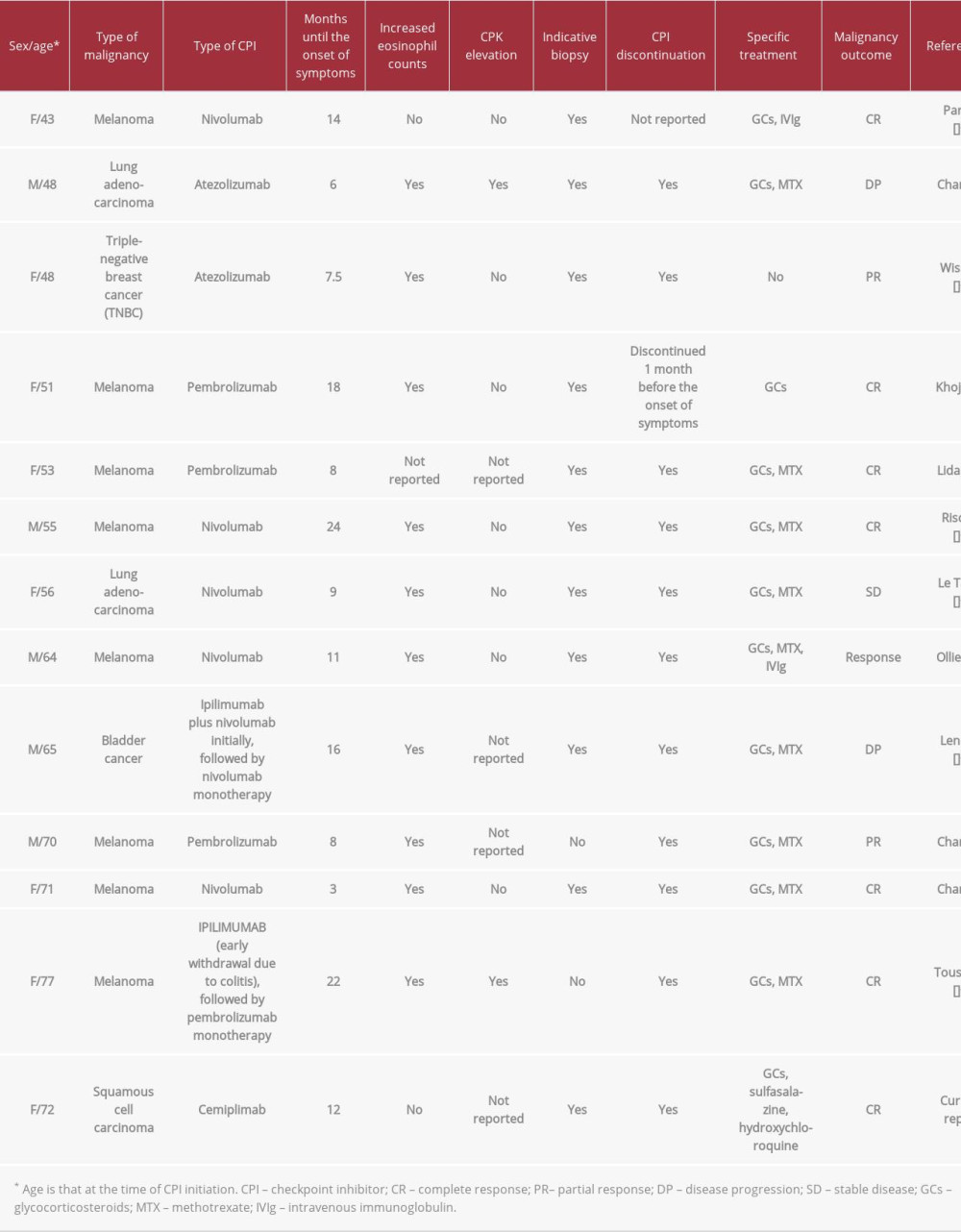
CPI-induced EF has been described in several other case reports (Table 1) [20–29]. Other implicated CPIs include nivolumab, pembrolizumab, and ipilimumab. To the best of our knowledge, our case is the first to describe cemiplimab-induced EF.
The mechanism of pathogenesis of CPI-induced EF remains to be established. It has been hypothesized that persistent activation of T and B cells may induce secretion of cytokines that promote eosinophil hyperactivation and the release of transforming growth factor beta; this, in turn, may lead to increased expression of type I collagen and other substances implicated in the classic manifestations of EF [20]. However, further investigation on the relationship between CPIs and EF is warranted.
Methotrexate and corticosteroids have emerged as the first-line treatments for EF; the choice of which agent to use can vary depending on individual patient characteristics [30]. Our patient refused methotrexate because of concern for developing a second malignancy. Therefore, an alternative regimen of corticosteroids, hydroxychloroquine, and sulfasalazine was selected. As of this writing, each of these drugs has been used for the treatment of EF, but there are no definitive data to support this 3-agent therapy.
Conclusions
EF is a rare adverse effect of immune CPIs characterized by woody induration of the skin and arthralgias. Diagnosis can often be established based on clinical presentation. However, a skin biopsy can be required in some individuals. Although EF is seldom fatal, prompt recognition and treatment is essential to reduce pain and optimize patient quality of life. There are no clinical guidelines for the management of immune checkpoint inhibitor-associated EF, but a regimen of immunosuppressant medications, which can include methotrexate and/or corticosteroids, and temporary cessation of the offending agent may control symptoms and prevent progression.
Figures
References:
1.. Shulman LE, Diffused fasciitis with hypergammaglobulinemia and eosinophilia. A new syndrome: J Rheumatic, 1974; 1(Suppl.); 46
2.. Falange V, Medgar TA, Frequency, levels, and significance of blood eosinophilia in systemic sclerosis, localized scleroderma, and eosinophilic fasciitis: J Am Acad Dermatol, 1987; 17; 648
3.. Shulman LE, Diffuse fasciitis with eosinophilia: A new syndrome?: Trans Assoc Am Physicians, 1975; 88; 70
4.. Kikuchi O, Murali H, Ikezoe K: Rinsho Shinkeigaku, 2004; 44; 299-302 [in Japanese]
5.. Belot V, Mulleman D, Perrinaud A: Ann Dermatol Venereol, 2007; 134; 673-77
6.. Diaz-Perez JL, Zubizarreta J, Gardeazabal J, Goday J, Familial eosinophilic fascitis induced by toxic oil: Med Cutan Ibero Lat Am, 1988; 16; 51-58
7.. Smith JD, Chang KL, Gums JG, Possible lansoprazole-induced eosinophilic syndrome: Ann Pharmacother, 1998; 32; 196-200
8.. Lucena Marotta F, Sanz Moreno J, Herrera Serrano L, Eosinophilic fasciitis: Its relationship with L-tryptophan ingestion: An Med Interna, 1995; 12; 337-39
9.. Hur JW, Lee HS, Uhm WS, Eosinophilic fasciitis associated with auto-immune thyroiditis: Korean J Intern Med, 2005; 20; 180-82
10.. Endo Y, Tamura A, Matsushima Y, Eosinophilic fasciitis: Report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome: Clin Rheumatol, 2007; 26; 1445-51
11.. Pardoll DM, The blockade of immune checkpoints in cancer immunotherapy: Nat Rev Cancer, 2012; 12(4); 252-64
12.. Lee A, Duggan S, Deeks ED, Cemiplimab: A review in advanced cutaneous squamous cell carcinoma: Drugs, 2020; 80(8); 813-19
13.. Migden MR, Rischin D, Schmults CD, PD-1 blockade with cemiplimab in advanced cutaneous squamous cell carcinoma: N Engl J Med, 2018; 379(4); 341-51
14.. Cappelli LC, Gutierrez AK, Bingham CO, Shah AA, Rheumatic and musculoskeletal immune-related adverse events due to immune checkpoint inhibitors: A systematic review of the literature: Arthritis Care Res, 2017; 69(11); 1751-63
15.. Abdel-Wahab N, Suarez-Almazor ME, Frequency and distribution of var-ious rheumatic disorders associated with checkpoint inhibitor therapy: Rheumatol (United Kingdom), 2019; 58; vii40-48
16.. Melissaropoulos K, Klavdianou K, Filippopoulou A, Rheumatic manifestations in patients treated with immune checkpoint inhibitors: Int J Mol Sci, 2020; 21(9); 3389
17.. Pinal-Fernandez I, Selva-O’Callaghan A, Grau JM, Diagnosis and classification of eosinophilic fasciitis: Autoimmun Rev, 2014; 13(4–5); 379-82
18.. Boin F, Hummers LK, Scleroderma-like fibrosingdisorders: Rheum Clin Disord, 2009; 34(1); 199
19.. Jinnin M, Yamamoto T, Asano Y, Diagnostic criteria, severity classification and guidelines ofeosinophilic fasciitis: J Dermatol, 2018; 45(8); 881-90
20.. Parker MJS, Roberts ME, Lorigan PC, Autoimmune fasciitis triggered by the anti-programmed cell death-1 monoclonal antibody nivolumab: BMJ Case Rep, 2018; 2018; 3-6
21.. Chan KK, Magro C, Shoushtari A, Eosinophilic fasciitis following checkpoint inhibitor therapy: four cases and a review of literature: Oncologist, 2020; 25(2); 140-49
22.. Wissam Y, Belcaid L, Wittoek R, Eosinophilic fasciitis in a patient treated by atezolizumab for metastatic triple-negative breast cancer: J Immunother Precis Oncol, 2019; 2(3); 101
23.. Khoja L, Maurice C, Chappell M, Eosinophilic fasciitis and acute encephalopathy toxicity from pembrolizumab treatment of a patient with metastatic melanoma: Cancer Immunol Res, 2016; 4(3); 175-78
24.. Lidar M, Giat E, Garelick D, Rheumatic manifestations among cancer patients treated with immune checkpoint inhibitors: Autoimmun Rev, 2018; 17(3); 284-89
25.. Rischin A, Brady B, McLean C, Ostor AJK, Immune checkpoint inhibitor-induced lymphocytic fasciitis: Intern Med J, 2018; 48(12); 1550-52
26.. Le Tallec E, Ricordel C, Triquet L, An original case of an association of eosinophilic fasciitis with cholangitis induced by nivolumab: J Thorac Oncol, 2019; 14(1); e13-15
27.. Ollier N, Tournier E, Meyer N, Nivolumab-induced eosinophilic fasciitis: A case report: Rheumatol Adv Pract, 2020; 4(1); 1-3
28.. Andrés-Lencina JJ, Burillo-Martínez S, Aragó , Miguel R, Eosinophilic fasciitis and lichen sclerosus in a patient treated with nivolumab: Australas J Dermatol, 2018; 59(4); e302-4
29.. Toussaint F, Hammon M, Erdmann M, Checkpoint inhibitor-induced eosinophilic fasciitis following high eosinophilia associated with complete response: Rheumatol (United Kingdom), 2019; 58(10); 1875-77
30.. Mazori DR, Femia AN, Vleugels RA, Eosinophilic fasciitis: An updated review on diagnosis and treatment: Curr Rheumatol Rep, 2017; 19(12); 74
Figures
In Press
06 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942937
12 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943244
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943275
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943411
Most Viewed Current Articles
07 Mar 2024 : Case report
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250