06 October 2021: Articles 
Hemophagocytic Lymphohistiocytosis Triggered by Leishmaniasis: A Case Report and Literature Review
Challenging differential diagnosis, Diagnostic / therapeutic accidents, Rare disease
Stefka Neycheva1ABDEF*, Boycho Oparanov1BE, Adriana Kamburova1AE, Rositsa Karalilova2AE, Vera Stoeva3AEDOI: 10.12659/AJCR.933012
Am J Case Rep 2021; 22:e933012






Abstract
BACKGROUND: Hemophagocytic lymphohistiocytosis (HLH) is a rare syndrome. It is a result of uncontrolled hyperactivation of the reticuloendothelial system with a release of a huge amount of proinflammatory cytokines in the bloodstream, causing a cytokine storm. It may be primary due to genetic defects and secondary, triggered by viruses, bacteria, parasites, lymphoproliferative, and autoimmune diseases. We present a rare case of HLH triggered by leishmaniasis. HLH accounts for only about 1% of all leishmaniasis cases.
CASE REPORT: The patient was a 40-year-old previously healthy woman, who has been diagnosed with secondary HLH caused by leishmaniasis. A chronic Epstein-Barr virus (EBV) infection was initially mistakenly interpreted as a trigger agent, because Leishmania amastigotes, present on the first bone marrow biopsy, were not recognized. The treatment with cyclosporin A and corticosteroids suppressed the cytokine storm and prevented the development of complications. Two months later, because of a reactivation of the disease, the patient was referred to a hematologist. A second bone marrow aspiration was performed, in which numerous Leishmania parasites were finally identified.
CONCLUSIONS: The aim of this case report is to provide more information about the rare phenomenon of secondary HLH triggered by leishmaniasis. Early recognition of the syndrome and its triggering agents will improve disease outcomes and prevent unnecessary treatment with immunosuppressive drugs.
Keywords: Leishmaniasis, Lymphohistiocytosis, Hemophagocytic, Epstein-Barr Virus Infections, Female, Herpesvirus 4, Human, Humans
Background
Leishmaniasis is caused by an obligate intramacrophage protozoa, and 21 Leishmania species have been identified to be pathogenic to humans [1]. The incidence of leishmaniasis in Europe is relatively low, ranging from 0.02/100 000 to 0.49/100 000 (and up to 8.53/100 000 in Turkey) [2].
There are 3 types of clinical presentations: cutaneous, mucosal, and visceral leishmaniasis. In 1% of all cases, leishmaniasis causes a secondary HLH syndrome.
HLH can occur in all age groups, causing death in 80% of cases. The most common triggering agents are bacterial and viral infections, and malignancies (HLH syndrome) [3]. The hypercytokinemia caused by autoimmune/auto-inflammatory conditions is called macrophage-activation syndrome (MAS).
The exact pathogenesis of the HLH induced by leishmaniasis and its role in parasite survival are unclear. Due to impaired control of infections, the parasites cause hyperactivation of the macrophages or histiocytes and they do not recognize normal and intact blood cells [4].
We report the case of a patient who presented with constitutional symptoms and septic fever. The poor clinical presentation and non-recognition of the protozoa on the first bone marrow biopsy delayed the diagnosis of leishmaniasis and treatment of the cause.
Case Report
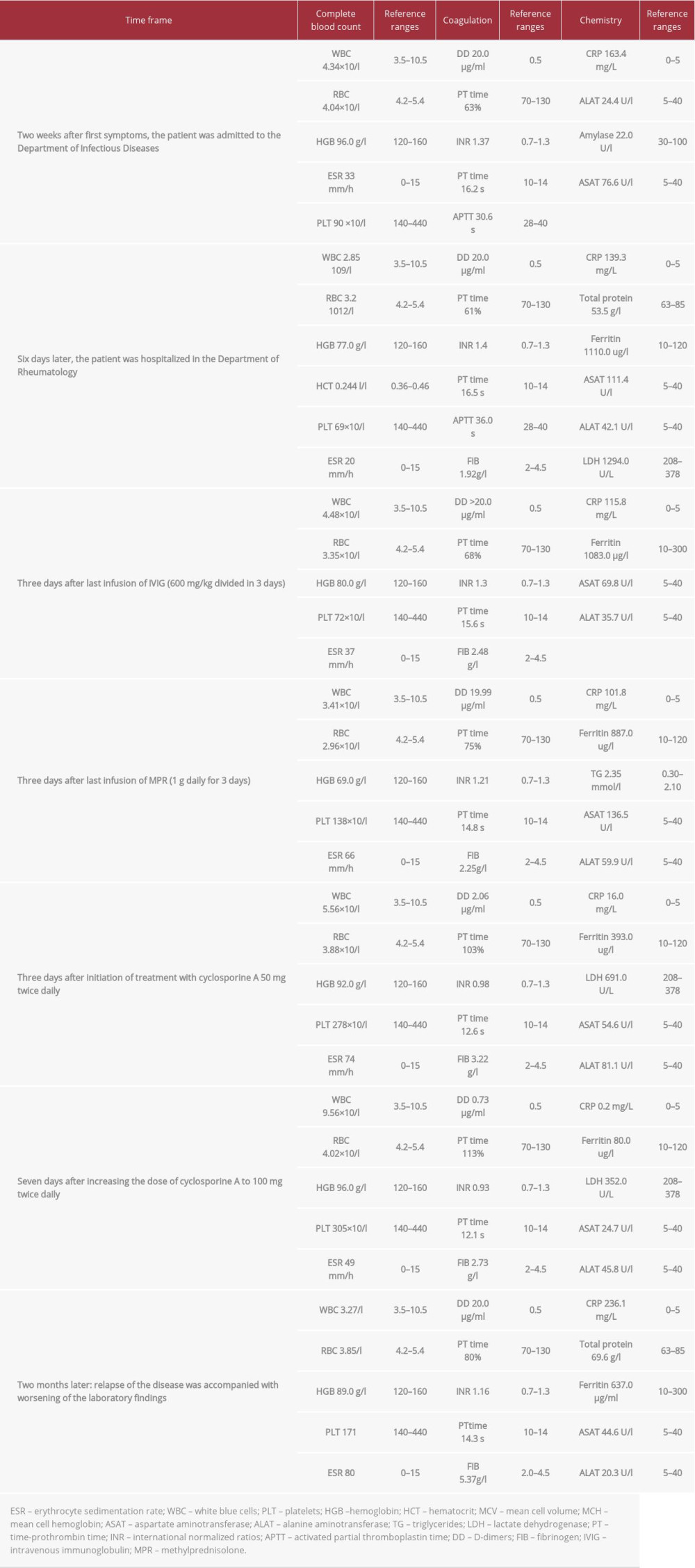
A 40-year-old, previously healthy woman presented with a history of fever up of to 40°C, followed by a gradual onset of fatigue, myalgia, profuse sweating, and chills for 2 weeks. Her family history was unremarkable. There was no information about co-existing diseases in the patient. She had no history of tobacco smoking or alcohol and substance abuse. She was hospitalized in the Department of Infectious Diseases with a diagnosis of observation sepsis (elevated acute-phase reactants, elevated D-dimers, septic fever) (Table 1).
Any active infections that could have caused her symptoms were excluded. Therefore, she was referred for a rheumatology consultation 6 days later with progressive pancytopenia and constitutional symptoms and suspected systemic lupus erythematosus due to polyserositis.
In the Department of Rheumatology, she presented with a fever of up to 40°C, fatigue, myalgia, profuse sweating, and chills. On physical examination, she appeared to be alert and aware of place and time. She had a fever of up to 38.4°C. The skin was pale and clammy. A lung examination showed tachypnea with a respiratory rate of 24 breaths per minute. The cardio-vascular function indicated tachycardia with a frequency of 105 beats per minute, regular rhythm; there were no murmurs, rubs, or clicks. Blood pressure was 90/60 mmHg. The abdomen was flat, soft, and non-tender, with physiological bowel sounds. The CNS and the peripheral nervous system (PNS) were functioning normally. She had normal mental status, muscle strength and power, and normal reflexes.
The chest X-ray was normal. The CT scan of the abdomen showed splenomegaly with a lesion considered as a possible infarction in the upper region. The CT scan also showed bilateral pleural effusions and pericarditis without lymphadenopathy. The transthoracic echocardiography was normal.
The findings in the bone marrow biopsy were interpreted by the histopathologist as reactive changes (Figure 1). Bone marrow samples were evaluated by the parasitologist, who concluded there was no evidence of parasitic disease.
ELISA EBV IgG against viral capsid antigen (VCA) was >>750 U/mL, and EBV IgM was in the “gray zone”. Polymerase chain reaction (PCR) for EBV was negative. All other tests for active infections concerning her condition were negative. ANA immunoblot test was negative.
After excluding infections and autoimmune and malignant diseases, we decided that the patient met the criteria for secondary HLH
Treatment was initiated with MPR 3.0 g IV, divided into 3 days (1 g daily) followed by normal human immunoglobulin 600 mg/kg divided into 3 days. After several new episodes of hemophagocytosis, treatment was started with cyclosporine A 50 mg twice daily, and MPR 60 mg IV once daily. Three days later, the dose of cyclosporin A was increased to 100 mg twice daily, which resolved all laboratory abnormalities and clinical symptoms.
After 2 months without any problems, the patient presented again to the Department of Rheumatology with sudden-onset adynamia, tachycardia, fever up to 40°C, and profuse sweating during the night. The physical examination revealed tachycardia of 120 beats per minute, hepatosplenomegaly, and subicteric skin. Hematologic abnormalities were found, similar to those described earlier (Table 1).
Because of the reactivations of HLH, we referred the patient to Hematology for treatment with etoposide and an assessment of the need of hematopoietic stem-cell transplantation (HSCT). They performed a second sternal puncture, where numerous Leishmania parasites were noted (Figures 2, 3).
Due to the established leishmaniasis, treatment with cyclosporine A was discontinued. The patient was started on the antileishmanial agent meglumine antimoniate. One week after the start of the etiologic treatment, the clinical and laboratory abnormalities were resolved.
Discussion
It is recommended that the diagnosis of HLH in adults be based on the HLH-2004 diagnostic criteria in conjunction with clinical judgment and the patient’s history [5]. The HLH-probability calculator (HScore) is a web-based online calculator that may be used as a helpful diagnostic tool in adults [6].
The typical clinical presentations of HLH include fever, hepatomegaly and/or splenomegaly, lymphadenopathy, typical skin rash, and laboratory constellation abnormalities [7], hemiparesis, aphasia, seizures, ataxia, coma, meningism, brain stem symptoms, or somnolence [8,9]. Lung symptoms include cough, dyspnea, and pleuritis. Interstitial infiltrates and nodules and ground-glass opacities can be seen on chest X-rays and computed tomography (CT) scans [10].
In the presented case, the poor clinical presentation, septic fever, and elevated acute-phase reactants required an exclusion of several conditions: infections, infectious endocarditis, sepsis, and lymphoproliferative and autoimmune diseases.
All serology tests for active infections, including leishmaniasis, were negative. EBV IgG against viral capsid antigen (VCA) was >>750 U/mL and EBV IgM in the “gray zone”.
Bone marrow samples were evaluated by the parasitologist. Due to the rarity of the condition and probably a lack of experience, the
Other life-threatening infections, such as malaria and brucellosis, were excluded [11,12].
A normal transthoracic echocardiography performed on the 14th day after admission excluded the possibility of an infectious endocarditis.
Because a pancytopenia developed rapidly in the course of the hospitalization, it was important to exclude a septic condition and lymphoproliferative disease.
Sepsis can cause a toxic suppression of the bone marrow with pancytopenia. The discrepancy between normal ESR and elevated CRP, normal procalcitonin levels, negative hemocultures, and episodes of hemolysis followed by hyperpyrexia do not support the diagnosis of sepsis.
A bone marrow biopsy did not confirm a lymphoproliferative disorder.
Because of the pancytopenia, polyserositis, and constitutional symptoms, we must keep in mind autoimmune conditions such as SLE, rheumatoid arthritis (RA), and vasculitis. Also, the patient did not fulfill the diagnostic criteria for either SLE or vasculitis (microscopic polyangiitis, granulomatosis with polyangiitis), which are the most common triggers for MAS syndrome. The absence of arthritis, morning stiffness, and negative rheumatoid factor (RF) ruled out an inflammatory arthritis.
After excluding infections and autoimmune and malignant diseases, we decided that the patient met the criteria for secondary HLH
Two treatment protocols were developed by the Histiocyte Society in 1994 [13] and 2004 [14]. The treatment of HLH requires an individualized approach, in accordance with the trigger factor [15].
Considering the pathophysiology of HLH triggered by a re-activated chronic EBV infection, we initiated a treatment with MPR 3.0 g IV, divided into 3 days (1 g daily) followed by normal human immunoglobulin 600 mg/kg divided into 3 days. After several new episodes of hemophagocytosis, treatment was started with cyclosporine A 50 mg twice daily and MPR 60 mg IV once daily. Three days later, the dose of cyclosporin A was increased to 100 mg twice daily, which led to resolution of all laboratory abnormalities and clinical symptoms.
One of the most severe complications of HLH, with a high mortality rate, is a diffuse intravascular coagulation (DIC). It is seen in 13.6% of patients with HLH [16]. Our patient developed coagulopathy, which could explain the normal ESR. This discrepancy between elevated ferritin, CRP, and normal ESR is very typical for HLH syndrome complicated by coagulation abnormalities (Table 1).
Owing to the timely application of cyclosporine A in the present case, the patient did not develop severe coagulation abnormalities or DIC syndrome.
Because of the reactivations of HLH after 2 months without problems, we referred the patient to Hematology for treatment with etoposide and assessment of the need for hematopoietic stem-cell transplantation (HSCT). They performed a second sternal puncture, where numerous Leishmania parasites were noted (Figures 2, 3). Another revision of the first bone marrow biopsy was performed by the same hematology team, where the parasites were also recognized.
The criterion standard for the diagnosis of the leishmaniasis is identifying the parasites in the relevant tissues aspirates or biopsies such as bone marrow, spleen, lymph nodes, or liver, skin slit smears or biopsies [17], but the sensitivity of the method is low.
Our literature review found 23 cases of Leishmania-related HLH reports in adults over the period of 7 years (2014–2020). One case of HLH caused by leishmaniasis in a child, who was initially diagnosed with EBV-triggered HLH, was reported by Singh et al [18], with findings very similar to those in the present report. In Bulgaria, for the period 1988–2011, there were 13 cases of leishmaniasis [19]. To the best of our knowledge, this is the first adult case of HLH triggered by Leishmania to be reported in Bulgaria.
Conclusions
Leishmaniasis and the HLH syndrome caused by it can present with a broad spectrum of atypical signs and symptoms. The scarce clinical presentation, wide differential diagnosis, and rarity of both diseases make them a diagnostic challenge for all clinical specialists. The aim of this case report is to reveal more information about the rare phenomenon of secondary HLH triggered by leishmaniasis. Timely recognition of the causative agent and early etiological treatment will prevent worsening of the initiated HLH and development of life-threatening CNS complications and DIC syndrome.
References:
1.. Singh S, New developments in diagnosis of leishmaniasis: Indian J Med Res, 2006; 123(3); 311-30
2.. Dujardin J-C, Campino L, Cañavate C, Spread of vector-borne diseases and neglect of leishmaniasis, Europe: Emerg Infect Dis, 2008; 14(7); 1013-18
3.. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Adult haemophagocytic syndrome: Lancet, 2014; 383(9927); 1503-16
4.. Morimoto A, Uchida K, Chambers JK, Hemophagocytosis induced by Leishmania donovani infection is beneficial to parasite survival within macrophages: PLoS Negl Trop Dis, 2019; 13(11); e0007816
5.. La Rosée P, Horne A, Hines M, Recommendations for the management of hemophagocytic lymphohistiocytosis in adults: Blood, 2019; 133(23); 2465-77
6.. Fardet L., HScore for Reactive Hemophagocytic Syndrome: MDCalc, 2020 [Cited 2021 Apr 20]https://www.mdcalc.com/hscore-reactive-hemophagocytic-syndrome
7.. Zhang Z, Wang J, Ji B, Clinical presentation of hemophagocytic lymphohistiocytosis in adults is less typical than in children: Clinics (Sao Paulo), 2016; 71(4); 205-9
8.. Pastula DM, Burish M, Reis GF, Adult-onset central nervous system hemophagocytic lymphohistiocytosis: A case report: BMC Neurol, 2015; 15; 203
9.. Haddad E, Sulis M, Jabado N, Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis: Blood J, 1997; 89(3); 794-800
10.. Seguin A, Galicier L, Boutboul D, Pulmonary involvement in patients with hemophagocytic lymphohistiocytosis: Chest, 2016; 149(5); 1294-301
11.. , About Malaria: CDC 24/7: Saving Lives, Protecting People, 2020 Available from: [cited 2021 Apr 20]https://www.cdc.gov/malaria/about/disease.html
12.. Bush L, Vazquez-Pertejo M.: Brucellosis – infectious diseases – MSD manual professional edition, 2021 . [cited 2021 April 5]https://www.msdmanualscom/professional/infectious-diseases/gram-negative-bacilli/brucellosis
13.. Henter JI, Samuelsson-Horne A, Arico M, Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation: Blood, 2002; 100(7); 2367-73
14.. Henter JI, Horne A, Aricó M, HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2007; 48(2); 124-31
15.. La Rosée P, Horne A, Hines M, Recommendations for the management of hemophagocytic lymphohistiocytosis in adults: Blood, 2019; 133(23); 2465-77
16.. Li J, Wang Q, Zheng W, Hemophagocytic Lymphohistiocytosis: Clinical analysis of 103 adult patients: Medicine (Baltimore), 2014; 93(2); 100-5
17.. Singh S, Sivakumar R, Recent advances in the diagnosis of leishmaniasis: J Postgrad Med, 2003; 49(1); 55-60
18.. Singh G, Shabani-Rad MT, Vanderkooi OG, Leishmania in HLH: A rare finding with significant treatment implications: J Pediatr Hematol Oncol, 2013; 35(3); e127-29
19.. Harizanov R.: Natural-focal, epidemiological, clinical-diagnostic and therapeutic aspects of visceral leishmaniasis in Bulgaria [Internet], 2012, Sofia, Bulgaria, National center of infectious and parasitic diseases [cited 2021 Jan 10]https://www.ncipd.org/images/UserFiles/File/D-r%20Harizanov/AVTOREFERAT.pdf
Figures
In Press
22 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943346
24 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943560
26 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943893
27 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942126
Most Viewed Current Articles
07 Mar 2024 : Case report 
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report 
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250