25 November 2021: Articles 
A Case of Paroxysmal Cold Hemoglobinuria Possessing Moderate Paroxysmal Nocturnal Hemoglobinuria-Type Erythrocytes
Challenging differential diagnosis
Takeshi Sugimoto1E*, Eri Masui2B, Shinya Ohata3B, Hideaki Goto1D, Takako Tomita2C, Hiromi Hashimoto2F, Yoshihiro Bouike4CDOI: 10.12659/AJCR.933102
Am J Case Rep 2021; 22:e933102






Abstract
BACKGROUND: Paroxysmal cold hemoglobinuria (PCH) is an autoimmune hemolytic disease caused by the Donath-Landsteiner (DL) antibody. Paroxysmal nocturnal hemoglobinuria (PNH) is a non-autoimmune hemolytic disease that is caused by a dysfunction in the synthesis of the glycosyl phosphatidylinositol anchor protein, resulting in the deregulation of the complement cascade and hypersensitivity for a hemolytic attack against erythrocytes. The mechanisms of these 2 hemolytic diseases are distinct. If PCH and PNH coexist in a patient, it is difficult to perform a differential diagnosis. We introduce a case of PCH that had DL antibodies and large PNH-type clones.
CASE REPORT: An 82-year-old female patient was referred to our hospital because of anemia. Initial workup revealed a negative antiglobulin test and a positive DL test. For the differential diagnosis, we surveyed the population of cells that had PNH-type clones, which revealed erythrocyte PNH clones (19.6%) and granulocyte PNH clones (73.3%). During the patient’s clinical course, mild hemolysis persisted without any attack. The percentage of the PNH-type erythrocytes was not obviously changed, and the DL antibody was detected 8 months after the initial admission. We determined that the persistent mild anemia was caused by concomitant diseases of PCH and PNH, although determining which of the 2 hemolytic systems was primarily responsible for the anemia was difficult.
CONCLUSIONS: When considering the differential diagnosis for hemolytic diseases, an adequate combination of laboratory tests for hemolysis is required.
Keywords: CD55 Antigens, CD59 Antigens, Hemoglobinuria, Paroxysmal, Aged, 80 and over, Anemia, Hemolytic, Autoimmune, Diagnosis, Differential, Erythrocytes, Female, Hemolysis, Humans
Background
Two types of diseases related to anemia are primarily associated with the cold type of autoimmune hemolysis: cold agglutinin disease (CAD) and paroxysmal cold hemoglobinuria (PCH). The causes of these 2 diseases are independent from each other. CAD occurs when cold agglutinin increases, which is due to the IgM class antibody. CAD is known as a complication of infections, such as mycoplasma, Epstein-Barr virus, legionella, or malignant lymphoma. However, CAD can also be idiopathic. PCH is caused by a biphasic autoantibody, the Donath-Landsteiner (DL) antibody, which is classified as an IgG. This autoantibody specifically binds to the P blood group antigen [1].
Paroxysmal nocturnal hemoglobinuria (PNH) is a hemolytic disease that manifests as anemia, thrombosis, bone marrow failure, and renal dysfunction. PNH occurs due to a disability in the synthesis of the glycosyl phosphatidylinositol (GPI) anchor portion. PNH lacks all, or parts of, the
The mechanisms of the above hemolytic diseases are distinct, and a differential diagnosis that leads to adequate treatment is important. The differential diagnosis of hemolytic diseases is based on clinical symptoms and the results of laboratory tests, including DAT or the indirect antiglobulin test (IAT), cold agglutinin titer, DL test, and specificity of erythrocyte antibody. The diagnostic surveillance for background diseases, such as lymphoma, myelodysplastic syndromes, collagen disease, and viral or bacterial infections, is also important. Here, we report a case of PCH possessing massive PNH-type clone cells.
Case Report
An 82-year-old female patient was referred to our hospital in February 2020 with anemia. Her anemia had been noted approximately 6 months before the referral. She had a previous history of surgery for appendicitis at 18 years of age and osteoporosis from around 60 years of age. She had been regularly visiting the family doctor for bronchiectasis. She had a pollen allergy and no transfusion history. On physical examination, her skin and conjunctiva were pale, without jaundice. Course crackles were found on lung auscultation. Slight edema was noted in the lower limbs, and no purpura was observed.
The color of the urine was dark yellow. Urine chemistry was negative for sugar and protein, 1+ for occult blood, and 1+ for urobilinogen. Urine microscopy showed red blood cells of 1–4/HPF and white blood cells of 2–3/HPF. A complete blood count showed the following: white blood cells, 9060/μL; red blood cells, 247×104/μL; hemoglobin, 7.7 g/dL; hematocrit, 24.5%; platelet count, 13.0×104/μL; reticulocyte count, 12.5×104/μL (reference range; 2–8×104/μL); and reticulocyte count percentage, 5.1% (range; 0.5–1.5%). The serological test showed some abnormalities: lactate dehydrogenase, 664 IU/L (range, 119–229 IU/L); total bilirubin, 0.72 mg/dL (range, 0.2–1.3 mg/dL); haptoglobin, <10 mg/dL (range, 50–220 mg/dL), with no hepatic or renal dysfunction. The immunological test showed the following: IgG, 1657 mg/dL (range, 870–1700 mg/dL); IgA, 393 mg/dL (range, 110–410 mg/dL); IgM, 281 mg/dL (range, 35–220 mg/dL); and IgE, 401 IU/mL (range, <358 IU/mL). The titer of complement CH50 was 49 U/mL (range; 32–56 U/mL); C3, 85 mg/dL (range, 65–135 mg/dL); and C4, 19 mg/dL (range, 13–35 mg/dL). The autoantibody test showed the following: anti-nuclear antibody titer, ×40; DNA antibody (RIA), <2.0 IU/mL (range, <6 IU/mL); and cold agglutinin titer, ×32 (range, <×64). All syphilis tests (treponemal test, rapid plasma reagin, and fluorescent treponemal antibody with absorption test) were negative. We tested for a parvovirus infection, and the results showed that the IgM antibody (EIA) was 0.26 (range, <0.8) and the IgG antibody (EIA) was 7.7 (range, <0.8), indicating a prior infection.
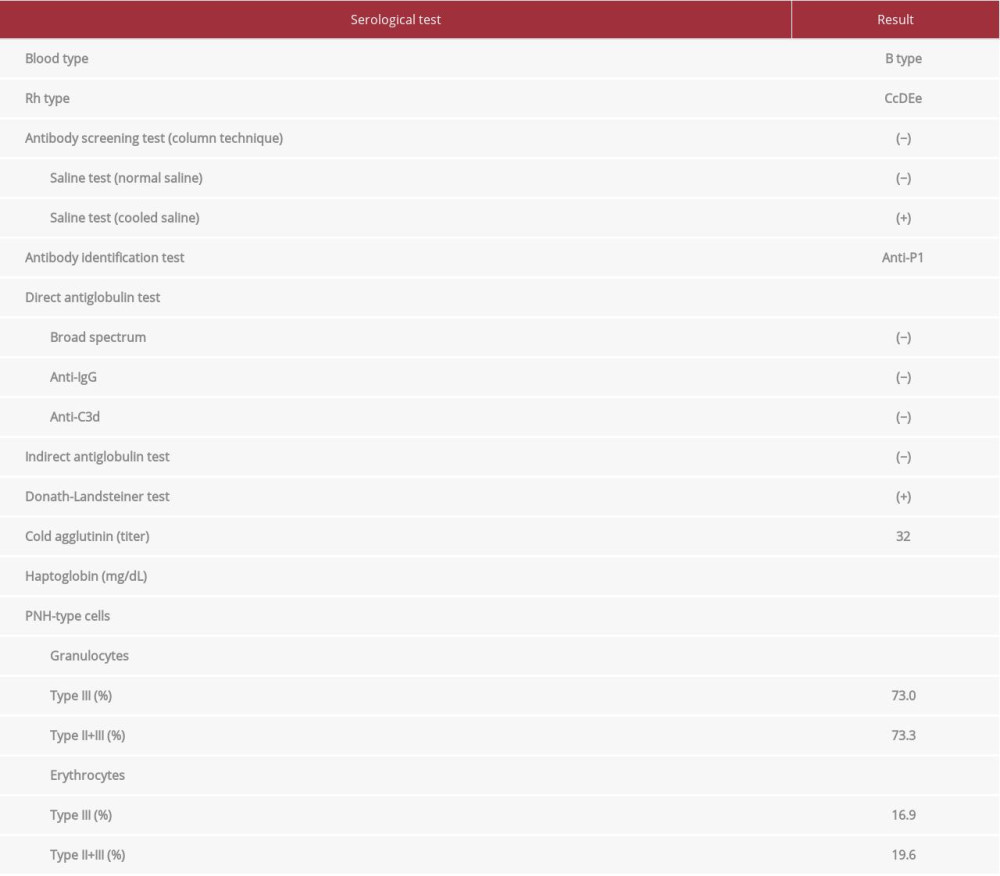
Blood typing showed type B and rhesus D-positive. The DAT (anti-IgG reagent and anti-C3d reagent) and IAT were negative. To rule out the cold type of autoimmune hemolytic anemia (AIHA), we performed the DL test, which was positive, indicating the existence of the cold-type autoantibody (Table 1). Further verification of the type of DL antibody was not performed. Erythrocytes of the p phenotype or Pk phenotype lack the P antigen, and if a patient’s sera contains anti-P antibody, it would not react with these erythrocytes. However, these rare erythrocytes were not available. The antibody identification test using Panel Cell® (Ortho-Clinical Diagnostics) and polyethylene glycol (Immucor Inc) IAT method with treatment of normal saline at room temperature showed a negative result. However, the above test with 4°C saline revealed an anti-P1 specific reaction. When the P1 blood group substance (#166129, Ortho) was treated with patient serum, the attenuation of P1-specific agglutination was observed, indicating that the patient’s serum contained an anti-P1 antibody in addition to the anti-P antibody. P1PK and globoside blood group testing revealed that her red blood cells had no P1 antigen (P2 blood type). The above results indicated that her anti-P1 antibody was not involved in hemolysis, and the DL antibody may have been related to hemolysis.
A chest computed tomography scan revealed bilateral bronchial wall thickening and bronchial dilation, which was consistent with bronchiectasis. No lymphadenopathy or tumor was found. Bone marrow aspiration analysis revealed a normocellular bone marrow with erythroblasts carrying mild megaloblastic changes (Figure 1A). No abnormalities were found in the megakaryocytes or granulocytes in the aspiration specimen. A flow cytometry analysis of the bone marrow aspiration showed a nonspecific pattern, and the karyotype was determined to be normal (46, XX). The bone marrow aspiration did not reveal any abnormalities, such as myelodysplastic syndromes or hematological malignancies, including malignant lymphoma. However, the flow cytometry analysis of the peripheral blood sample, focusing on the differential diagnosis of PNH, was performed by the outsourcing company SRL Inc (Tokyo, Japan). Flow cytometry analysis is useful for detecting the population of PNH-type cells. Briefly, peripheral blood cells were collected and 2-dimensional expansion with forward scatter and side scatter was performed. For analysis of erythrocytes, PE-labeled CD235a (glycophorin A) antibody was used as the erythrocyte lineage marker, and a cocktail reagent of FITC-labeled CD55 antibody and FITC-labeled CD59 antibody was used to detect erythrocyte PNH clones (direct fluorescent antibody method). We expressed erythrocyte PNH clones as the total of type II PNH erythrocytes (partial deficiency) and type III PNH erythrocytes (complete deficiency). For analysis of granulocytes, PE-labeled CD11b antibody was used as the granulocyte lineage marker, and fluorescein-labeled inactive toxin aerolysin antibody was used to detect granulocyte PNH clones. Aerolysin has the characteristic of binding specifically to GPI-anchored membrane proteins, including CD55 and CD59. The result showed the existence of distinct PNH clones (erythrocyte PNH clones, 19.6%; granulocyte PNH clones, 73.3%) (Figure 1B, 1C). We did not investigate mutation analysis of the PIG-A gene.
The patient was diagnosed with hemolytic anemia; however, the hemolytic activity was mild. She was transfused with 2 units of P1 antigen-negative red blood cells after admission. There were no hemolytic transfusion reaction complications. Patient follow-up after 8 months did not reveal any use of steroids, immunosuppressive drugs, or complement C5 inhibitors. Her hemolytic anemia gradually improved until June 2020. She needed to receive another 2 units of red blood cell transfusion in August owing to a low hemoglobin level of 6.7 g/dL, which was caused by mild persistent hemolysis and transient bronchopneumonia. Eight months later, we again performed flow cytometry analysis for the erythrocyte PNH clones, which revealed a population of 15.6%. The DL test was still positive at that point (Figure 2). During the clinical course, hypocomplementemia (C4, CH50) was not noted despite the persistence of the indices of hemolytic anemia, such as an increase in lac-tate dehydrogenase and hypohaptoglobulinemia at below the level of lower sensitivity.
Hyperbilirubinemia was not observed (maximum level of total bilirubin was 0.88 mg/dL in April 2000). We determined that her hemolytic anemia was caused by the concomitant diseases of PCH and PNH, and the degree of hemolysis was mild (Figure 2).
Discussion
The presented case of hemolysis had PCH characteristics of possessing a moderate clone of PNH-type cells. PCH is classified as a type of AIHA in which the DL antibody is mostly responsible for the disease. PCH is classified as 3 types: acute, chronic syphilitic, and chronic non-syphilitic [3]. Our patient had the chronic non-syphilitic type and, even though PCH itself is a rare disease, this type is frequently seen in older patients. PCH often appears in the winter season because erythrocytes in PCH show hypersensitization to cold exposure. The clinical features of PCH consist of the symptoms of intravascular hemolysis, such as anemia, jaundice, and hemoglobinuria. An increase in reticulocytes and a decrease in complement levels are also seen during laboratory testing [4]. Although there was no jaundice, hyperbilirubinemia, or hemoglobinuria in the patient, a decrease in haptoglobin level and increase in lactate dehydrogenase level was noted. The reason for the lacking part in the hemolytic index in this case may be due to the slow progression. Regarding the discrimination of the cold type of AIHA, CAD is a major disease that usually carries the high cold agglutinin titer and IgM class autoantibody [5]. In contrast, our patient showed a low titer level (×32) of CA and carried the DL autoantibody, which indicated that this case was not CAD but PCH. We surveyed whether there were background diseases in our patient, such as a hematological malignancy or collagen disease, and concluded this case was an idiopathic type of PCH without any background disease. In terms of DAT, monospecific DAT is usually negative for IgG but frequently positive for C3d fragment in patients with PCH [5]. However, some patients with PCH have had a negative DAT for the C3d fragment [4,6] and these patients tend to have the chronic type of PCH [7]. Our patient had a negative DAT for the C3d fragment, estimating that her DAT-negative status was recognized as the chronic non-syphilitic type of the elderly PCH.
However, PNH could have coexisted in our patient because PNH-type erythrocytes and granulocytes can be detected in a distinct population. PNH is a non-autoimmune hemolytic disease that occurs due to complement-mediated hemolysis. The hemolytic sensitivity for complement-mediated stimulation in PNH patients is increased owing to a lack of complement regulatory proteins, such as
While monitoring the level of anemia during the patient’s clinical course, the fraction of PNH-type erythrocytes did not significantly change and she had no hypocomplementemia, which may mean that mild hemolysis persisted without an attack. Considering that PNH-type erythrocytes seem to be more sensitive to immune-mediated stimulation than normal erythrocytes when an antigen-antibody reaction exists, it is difficult to determine which of the 2 diseases mainly contributed to the hemolysis. Kalam et al reported a concomitant AIHA and PNH case in which erythrocyte PNH clones were a much smaller population than were granulocyte PNH clones and hypothesized that erythrocyte PNH clones were lost by hemolysis [8]. In the present case, the population of granulocyte PNH clones (73.3%) was much larger than that of erythrocyte PNH clones (19.6%) at diagnosis, and this discrepancy in population became slightly increased during the clinical course. It is speculated that the hemolysis of erythrocyte PNH clones was promoted by sensitization of the DL antibody.
The anti-P1 and DL antibodies were detected in the blood of this patient by the irregular antibody detection test on erythrocytes. It is unknown which of the 2 antibodies was responsible for the hemolysis. The anti-P1 antibody of the patient reacted at low temperatures and did not react on the IAT. The anti-P1 antibody was neutralized with the P1 substance. The DL antibody is known to have specificity to the P antigen [11–13]. Since the P antigen is expressed in the P1 and P2 blood group types on red blood cells, the DL antibodies reacted with both types of erythrocytes. Basically, the P1 blood group has the P1, P, and PP1Pk antigens, and the P2 blood group has only the P and PP1Pk antigens on the P1PK and globoside blood group system. No relation exists between the anti-P1 antibody and PCH. A few cases of PCH showing an anti-P1 antibody reaction whose P blood group was the P1 type have been reported [13,14]. In the present case, we confirmed that this patient had the P2 blood group type (no existence of P1 antigen). Therefore, the anti-P1 antibody would not have caused hemolysis in our patient. We speculated that the DL autoantibody was responsible for her autoimmune hemolysis.
Conclusions
We investigated a PCH case possessing a moderate cloning of PNH-type cells. When considering the differential diagnosis for hemolytic diseases, an adequate combination of laboratory tests for hemolysis is required.
Figures
References:
1.. Levine P, Celano MJ, Falkowski F, The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H): Transfusion, 1963; 3; 278-80
2.. Lima M, Laboratory studies for paroxysmal nocturnal hemoglobinuria, with emphasis on flow cytometry: Pract Lab Med, 2020; 20; e00158
3.. Dacie JV, The hemolytic anemia, congenital and acquired. Part II: The autoimmune hemolytic anemias, 1962; 560, London, Churchill
4.. Shanbhag S, Spivak J, Paroxysmal cold hemoglobinuria: Hematol Oncol Clin North Am, 2015; 29; 473-78
5.. Berentsen S, Hill A, Hill QA, Tvedt THA, Michel M, Novel insights into the treatment of complement-mediated hemolytic anemias: Ther Adv Hematol, 2019; 10; 1-20
6.. Cooling LL, Kids, colds, and complement: paroxysmal cold hemoglobinuria: Transfusion, 2017; 57; 1332-35
7.. Sokol RJ, Booker DJ, Stamps R, Erythropoiesis: Paroxysmal cold haemoglobinuria: A clinico-pathological study of patients with a positive Donath-Landsteiner test: Hematology, 1999; 4; 137-64
8.. Kalam S, Beale R, Hughes D, Coombs-positive paroxysmal nocturnal haemoglobinuria: Oxf Med Case Rep, 2020; 3; 79-82
9.. Zupańska B, Bogdanik I, Fabijanska-Mitek J, Pyl H, Autoimmune haemolytic anaemia with a paroxysmal nocturnal haemoglobinuria-like defect: Eur J Haematol, 1999; 62; 346-49
10.. Mengel CE, Hyman BN, O’Malley BW, Howell DA, Coexistent paroxysmal nocturnal and cold hemoglobinuria preceded by aplastic anemia: A case report and family study: Blood, 1964; 24; 451-57
11.. Kilty M, Ipe TS, Donath-Landsteiner test: Immunohematology, 2019; 35; 3-6
12.. Worlledge SM, Rousso C, Studies on the serology of paroxysmal cold haemoglobinuria (P.C.H.), with special reference to its relationship with the P blood group system: Vox Sang, 1965; 10; 293-98
13.. O’Neill BJ, Marshall WC, Paroxysmal cold haemoglobinuria and measles: Arch Dis Child, 1967; 42; 183-86
14.. Wise SC, Tinsley SH, Cook LO, Paroxysmal cold hemoglobinuria: A case report: Immunohematology, 2012; 28; 118-23
Figures
In Press
16 Mar 2024 : Case report ")
Am J Case Rep In Press; DOI: 10.12659/AJCR.943214
16 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943010
16 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943687
17 Mar 2024 : Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.943070
Most Viewed Current Articles
07 Mar 2024 : Case report 
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report 
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250