29 December 2021: Articles 
Challenges of Managing Multiple Myeloma Patients with Sickle Cell Disease: A Case Report and Review of Literature
Diagnostic / therapeutic accidents, Educational Purpose (only if useful for a systematic review or synthesis), Rare coexistence of disease or pathology
Maroun Bou Zerdan1ABDEF*, Maria Julia Diacovo2EF, Chakra P. Chaulagain1ABCDEFDOI: 10.12659/AJCR.933470
Am J Case Rep 2021; 22:e933470






Abstract
BACKGROUND: A congenital hemolytic anemia, sickle cell disease can present with various clinical findings. Sickle cell disease is typically a disease of younger people and multiple myeloma typically occurs in older individuals. Multiple myeloma is rare among patients with sickle cell disease. Both multiple myeloma and sickle cell disease can cause various types of organ damage by different mechanisms.
CASE REPORT: We report a case of a patient who was born with sickle cell disease and presented with multiple myeloma later in life. Although he responded to anti-myeloma therapy, he died of hepatic and renal failure from complications of both multiple myeloma and sickle cell disease.
CONCLUSIONS: We discuss the complexity involved and present a review of the literature on managing multiple myeloma in relation to hepatic iron overload and end-stage renal disease in the setting of multiple myeloma and underlying sickle cell disease.
Keywords: Anemia, Sickle Cell, Multiple Myeloma, pomalidomide, Humans, Iron Overload, Male
Background
A few case reports have been published in which hematologic malignancies occurred in the setting of sickle cell disease (SCD) [1–6]. A congenital hemolytic anemia, sickle cell disease can present with various clinical findings. Hematologic malignancies, on the other hand, can present in different age groups depending on the subtype of malignancy. For example, multiple myeloma (MM), a malignancy of plasma cells, has a median age of onset between 66 and 70 years. Hematologic malignancy is a rare occurrence in sickle cell disease [7]. In the United States, the expected prevalence of sickle cell anemia is 1 in 625 (0.16%) persons at birth, and the lifetime risk of getting MM is 1 in 132 (0.76%) [8,9]. With that being said, the chance of having SCD and MM in the same patient is extremely low. One of the earliest reported cases of SCD in the setting of MM was by Anderson et al in 1975, in which extensive cone-plate viscometric studies were conducted. Interestingly, the patient had an increased frequency of vaso-occlusive crisis that occurred a few months preceding the diagnosis of MM. Further studies confirmed that sickled cells interacted with proteins that resulted from the hyperviscosity state of MM. An interaction can occur between the effects of SCD, which decreases red cell deformability, and MM, which augments whole-blood viscosity by increasing both plasma viscosity and the sickling phenomenon. The paraproteinemia associated with multiple myeloma and the associated change in bone marrow microenvironment can augment red blood cell sickling, leading to increased incidence of painful vaso-occlusive crisis in patients with MM and SCD. New-onset transfusion dependency and increased frequency of vaso-occlusive painful crisis were noted in our patient and in the cases reviewed in the literature when MM presented in a patient with SCD. We discuss the challenges associated with both the diagnosis and management of this rare clinical combination.
Case Report
In February 2018, a 70-year-old black man with a history of sickle cell disease (HbSS) since birth, necessitating blood transfusion since 2016, stage IV chronic kidney disease (CKD), chronic diastolic heart failure, hypertension, transfusional iron overload, and hyperlipidemia presented due to an increase in episodes of painful bone crises and increasing shortness of breath. Prior to 2016, his SSD was mild, and he rarely had painful vaso-occlusive bone crisis, did not have nephropathy, and never needed blood transfusion. Laboratory results were significant for red cell count 2.01 M/uL (4–5.5 M/uL), hemoglobin 6.7 g/dL (13.5–17.5 g/dL), hematocrit 19.6% (41–50%), mean cell volume (MCV) 97.5 fL (80–100 fL), ferritin 67 850 ng/mL (30–400 ng/mL), transferrin saturation 70% (15–50%), bilirubin 1.6 mg/dL (0.2–1.3 mg/dL), and creatinine 3.75 mg/dL (0.73–1.22 mg/dL). Despite receiving frequent blood transfusions along with deferoxamine for secondary transfusional iron overload, and worsening CKD, the cause of the clinical deterioration was never fully investigated until the patient switched his care to us due to changes in the medical insurance. On our initial evaluation, we were intrigued by the new-onset worsening anemia, increased need for transfusion, and increased frequency and severity of painful vaso-occlusive bone crises. This led to further investigation for potential causes of bone marrow hypoproliferative anemia, including MM, which yielded an IgA lambda (λ) monoclonal gammopathy with a serum IgA of 439 mg/dL (80–350 mg/dL), serum free kappa (κ), and λ of 20.2 mg/L (0.33–1.94) and 181.9 mg/L (0.4–4.2 mg/dL), respectively, with a ratio of 0.11 (0.26–1.65). A bone marrow aspiration and biopsy were done in March 2018, which revealed λ restricted malignant plasma cells with 50–60% bone marrow cellularity (Figure 1). The bone survey reported numerous lytic bone lesions without pathologic fractures. The worsening transfusion-dependent anemia, painful vaso-occlusive bone crises, and progressive CKD were attributed to MM and treatment was started with bortezomib, cyclophosphamide, and dexamethasone (CyBorD). The goals of therapy were achieving transfusion independence, as well as to protect, preserve, and potentially reverse progressive myeloma-associated nephropathy (myeloma kidney). To treat anemia in the setting of advanced CKD, darbepoetin alfa was initiated. The patient tolerated the CyBorD regimen with minimal adverse effects, and iron chelation therapy was held during chemotherapy with a plan of reinitiating it in the future. After 4 cycles, cyclophosphamide was discontinued due to elevation of hepatic enzymes, and lenalidomide at a renally-adjusted dose of 5 mg daily was added to bortezomib and dexamethasone. By August 2018, a repeat bone marrow biopsy reported <5% clonal plasma cells and serology was consistent with very good partial response (VGPR). The patient was felt not to be a candidate for autologous hematopoietic stem cell transplant (HSCT) in light of his comorbidities. He then developed peripheral neuropathy from bortezomib; therefore, therapy was further de-escalated to lenalidomide 5 mg every other day in light of renal failure advancing to stage V CKD at that point, and bortezomib was discontinued. Despite the response to anti-myeloma therapy, CKD progressed from stage IV to stage V (end-stage renal disease/ESRD) at 5 months after initiating anti-myeloma therapy. Iron chelation therapy was reintroduced. The patient never became transfusion-independent throughout the chemotherapy and continued to need RBC transfusion every 4 weeks on average. He had multiple hospitalizations for pneumonia, congestive heart failure exacerbations, and several episodes of painful vaso-occlusive crisis involving bones during the therapy.
In April 2019, the patient presented to the Emergency Department with new-onset jaundice and extreme fatigue. His bilirubin (0.1–1.2 mg/dL), ALT (7–55 U/L) and AST (8–48 U/L) had increased from baseline normal to 26.9 mg/dL, 97 U/L, and 154 U/L, respectively. A liver biopsy revealed significant iron deposits in hepatocytes and Kupffer cells with presence of bridging fibrosis and cirrhosis (Figure 2). Deferasirox was continued due to worsening renal failure, and the patient was started on hemodialysis. Hemodialysis improved hepatic encephalopathy briefly, but he developed respiratory distress. A CT scan of the thorax showed tree-in-bud pattern in the lungs and a right lung mass concerning for infection vs neoplastic process. The patient continued to deteriorate and died from septic shock from Citrobacter bacteremia originating from spontaneous bacterial peritonitis.
Discussion
LITERATURE REVIEW OF CO-OCCURRENCE OF SICKLE CELL DISEASE AND MULTIPLE MYELOMA:
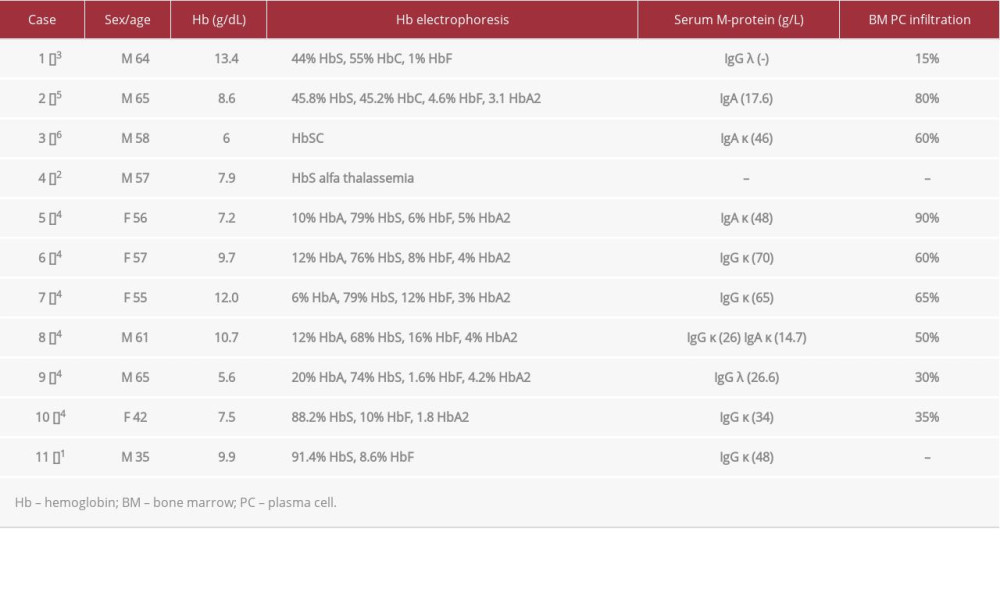
In this report, we present our case of a patient with HbSS SCD and MM along with a brief literature review of the available data of SCD and MM. To date, there are only 12 known cases of sickle cell anemia occurring concurrently with MM (Table 1). The earliest reported case, which was not included in the table due to insufficient information, dates to 1971.
Well-known causes of erythropoiesis-related hemolytic anemia include but are not limited to thalassemia, hereditary spherocytosis, sickle cell anemia, and congenital dyserythroblastic anemia [10–12]. When investigating these patients, malignant causes of anemia due to bone marrow involvement should also be considered. Some examples are primary malignancies, such as acute-chronic leukemias, plasma cell dyscrasias, Hodgkin and non-Hodgkin lymphoma, malignant histiocytosis, and secondary metastatic tumors from primary malignancies, such as breast cancer, renal medullary cancer, and hepatocellular cancer [1,2,13–35]. In most reported cases where the patient had SCD, the frequency and incidence of bone crises increased approximately 2 years ahead of clinical diagnosis of MM. This applies to our patient as well.
Since our report involves patients diagnosed with MM over a broad range of time, different treatment modalities have been applied. Out of the 12 reported cases, only 5 have reported the patient’s outcome, with 2/5 being eventual mortality at the time of publication [3,7]. Interestingly, one of these patients was a 39-year-old pregnant woman with sickle cell anemia who was found to have MM; she was the first case to be reported in the literature. Even though she proceeded to full-term delivery, she died 3 years 8 months later. The second patient with reported mortality was a 64-year-old man who received oral melphalan (0.25 mg/kg) and prednisone (2 mg/kg), local radiation, and CHOP (cyclophosphamide, Adriamycin, vincristine, and prednisone) chemotherapy. He died 2 years later with disseminated intravascular coagulation and Gram-negative sepsis; an autopsy revealed progression of MM involving various organs. As mentioned above, our patient’s treatment regimens included CyBorD and lenalidomide. Our patient’s MM responded well to CyBorD, achieving VGPR (Figure 3). The iron overload leading to hepatic cirrhosis and potentially cardiac hemosiderosis and related complications in the setting of SCD was the main contributor to mortality. On the other hand, the patients mentioned by Oyadini et al, Rodriguez et al, and Paydas were alive and treated with different modalities. The first received chemotherapy with melphalan, prednisolone, and thalidomide.
The second patient began therapy with melphalan, prednisolone, and zoledronic acid. The third received vincristine, doxorubicin, and dexamethasone before eventually receiving HSCT.
IS POMALIDOMIDE A BETTER IMMUNOMODULATORY DRUG THAN LENALIDOMIDE IN PATIENTS WITH SICKLE CELL DISEASE WITH MULTIPLE MYELOMA?:
Pomalidomide, a third-generation immunomodulatory drug used against MM, was proven to induce fetal hemoglobin production by inducing a fetal-like erythroid differentiation program [36]. This drug acted early by transiently delaying erythropoiesis at the burst-forming unit-erythroid/colony-forming unit-erythroid transition, but without affecting terminal differentiation [36]. In addition, patients treated with MM with pomalidomide demonstrated increased in vivo γ-globin levels in their erythrocytes [36]. Here it is worth comparing pomalidomide’s antitumor and immune-stimulating properties with lenalidomide, a second- generation immunomodulatory drug (IMID) frequently used against MM. Compared with lenalidomide, not only does pomalidomide have a different substrate degradation kinetics and increased potency against cereblon, but it also has a distinct gene activation profile [37]. In a study by Siegel et al, patients with relapsed/refractory multiple myeloma received pomalidomide plus low-dose dexamethasone immediately after lenalidomide-based treatment failure, reporting a median progression-free survival of 12.2 months and a median overall survival of 41.7 months, with minimal treatment-emergent adverse events. These findings support the use of pomalidomide-based therapy in lenalidomide-pretreated patients, including those who have become refractory to lenalidomide [38]. Together, these data reveal the molecular mechanisms by which pomalidomide reactivates fetal hemoglobin and is efficacious for patients who have become refractory to lenalidomide, reinforcing its potential use as a treatment for patients with MM and β-hemoglobinopathies [36]. This observation makes us consider whether pomalidomide should be used prior to lenalidomide in patients with SCD and MM, given the potential for pomalidomide to promote erythropoiesis.
Both lenalidomide and pomalidomide have teratogenic potential and are contraindicated in pregnancy.
MECHANISMS OF ORGAN DYSFUNCTIONS IN PATIENTS WITH MM AND SCD:
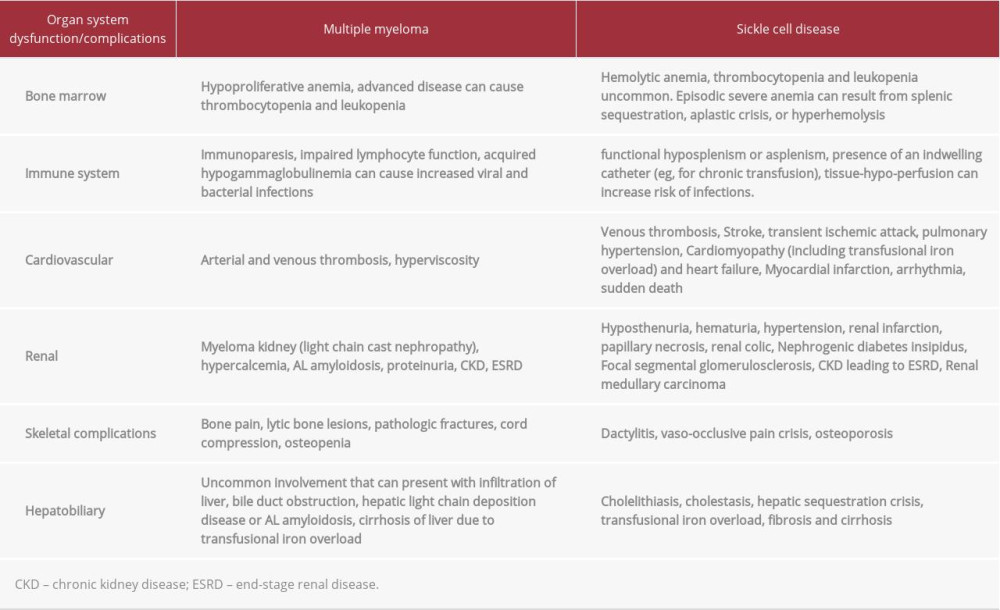
Both MM and SCD can cause multiple-organ system dysfunctions, as detailed in Table 2. As illustrated, the combination of MM and SCD can lead to rapidly progressive multiple-organ dysfunction and highlights the challenges of treating MM when it occurs in the setting of SCD. This is probably why SCD patients with MM are not usually HSCT candidates, which was also the case with our patient. For example, our patient had progression of CKD from stage IV to stage V (ESRD) despite very good partial response to anti-myeloma therapy. In general, an anti-myeloma response leads to improvement of myeloma-related renal insufficiency, but it was not the case in our patient. The progression to ESRD is likely multifactorial and may have been contributed to by transfusional iron overload, leading to congestive heart failure, and liver failure leading to renal ischemia from hepatorenal and cardiorenal syndrome, respectively. In addition, the possibility of renal AL amyloidosis cannot be entirely ruled out due to lack of renalbiopsy in this case.
Conclusions
SCD and MM is a rare clinical combination. SCD is a disease of younger patients and MM is a disease of older patients; therefore, the combination is uncommon. When a relatively mild SCD patient without prior history of transfusion becomes transfusion-dependent because of new-onset worsening anemia and develops new-onset bone pain and increased frequency and severity of painful bone vaso-occlusive crisis, a primary bone marrow hematologic malignancy such as multiple myeloma should be considered. The management is challenging, as one must deal with the dual challenges of MM and SCD affecting multiple organ systems, leading to rapid clinical deterioration. Anti-myeloma therapy can increase the need for transfusion due to suppression of erythropoiesis, and iron overload can get worse relatively faster, leading to heart and liver failure. Both MM and SCD can work together to cause renal dysfunction with progression of CKD, leading to ESRD.
Infectious complications are common in both SCD and MM, which further complicates therapy. Newer immunotherapy targeting CD38 (daratumumab and isatuximab) may be better tolerated and pomalidomide may have a potential therapeutic role in this setting. Little is known about the utility of HSCT in this patient population due to the paucity of data, as we found only 1 patient with SCD and MM treated with HSCT in the literature.
Figures
References:
1.. Anderson IS, Yeung K-Y, Hillman D, Lessin LS, Multiple myeloma in a patient with sickle cell anemia: Interacting effects on blood viscosity: Am J Med, 1975; 59(4); 568-74
2.. Paydas S, Sickle cell anemia and hematological neoplasias: Leuk Lymphoma, 2002; 43(7); 1431-34
3.. Stricker RB, Linker CA, Crowley TJ, Embury SH, Hematologic malignancy in sickle cell disease: Report of four cases and review of the literature: Am J Hematol, 1986; 21(2); 223-30
4.. Kaloterakis A, Filiotou A, Konstantopoulos K, Multiple myeloma in sickle cell syndromes: Haematologia (Budap), 2001; 31(2); 153-60
5.. Olaniyi JA, Shonde-Adebola KB, Immunoglobulin a myeloma in a newly diagnosed sickle cell disease patient: Int J Appl Basic Med Res, 2018; 8(3); 177-80
6.. Rodríguez LR, Estrada EE, Cabrera OÁ, Concurrent multiple myeloma, sickle-cell disease and diabetes mellitus: A case report: Revista de Hematología, 2012; 13(1); 36-38
7.. Talerman A, Serjeant GR, Milner PF, Normal pregnancy in a patient with multiple myeloma and sickle cell anaemia: West Indian Med J, 1971; 20(2); 97-100
8.. Motulsky AG, Frequency of sickling disorders in US blacks: N Engl J Med, 1973; 288(1); 31-33
9.. , Key Statistics About Multiple Myeloma Available from: https://www.cancer.org/cancer/multiple-myeloma/about/key-statistics.html
10.. Koudieh MS, Afzal M, Rasul K, Baez-Giangreco A, Intrathoracic extramedullary hematopoietic tumor in hemoglobin C disease: Arch Pathol Lab Med, 1996; 120(5); 504-6
11.. Chuang C, Chu S, Fang J, Wu J, Adrenal extramedullary hematopoietic tumor in a patient with beta-thalassemia: J Formos Med Assoc, 1998; 97(6); 431-33
12.. Kouraklis G, Dosios T, Intrathoracic extramedullary hematopoiesis simulating tumor, in a patient with sickle cell anemia: Eur J Cardiothorac Surg, 1994; 8(4); 220-21
13.. Salmassi S, Currie ET, Bolf EC, Management of Hodgkin’s disease in a patient with sickle cell anemia: Cancer, 1981; 48(2); 252-54
14.. Katz F, Reeves B, Alexander S, Leukaemia arising in donor cells following allogeneic bone marrow transplantation for β thalassaemia demonstrated by immunological, DNA and molecular cytogenetic analysis: Br J Haematol, 1993; 85(2); 326-31
15.. Pike M, Morrow R, Kisuule A, Mafigiri J, Burkitt’s lymphoma and sickle cell trait: Br J Prev Soc Med, 1970; 24(1); 39-41
16.. Russo A, Schiliro G, Thalassemia major and malignancies: Am J Hematol, 1987; 24(1); 111-12
17.. Fabris P, Coser P, Prinoth O, [Multiple myeloma terminating in acute myeloid leukaemia in a patient with a beta-thalassemia trait. A case report (author’s transl).]: Haematologica, 1977; 62(6); 629-36 [in Italia]
18.. Borgna-Pignatti C, De Stefano P, Hepatocellular carcinoma in thalassemia major: Med Pediatr Oncol, 1986; 14(6); 327-28
19.. Castoldi G, Grusovin G, Scapoli G, Spanedda R, Association of multiple haematological disorders (acute myeloblastic leukaemia, paraproteinaemia, and thalassaemia) in a 46, XX/46, XXqi female: Acta Haematol, 1971; 46(5); 294-306
20.. Schiliro G, Russo A, Marino S, Russo G, Occurrence of lymphoma with bone marrow involvement in a boy with β+ thalassaemia major: Clin Lab Haematol, 1979; 1(4); 325-28
21.. Balestrazzi P, Thalassemia and Hodgkin lymphoma: Blut, 1985; 50; 55
22.. Felici W, Ballati G, Vignetti M, Letter to the editor: Acute myeloid leukemia in a child affected by β-thalassemia major: Am J Hematol, 1988; 29(2); 124-24
23.. Lau B, Eife R, Lampert F, [Acute lymphoblastic leukemia after tuberculosis in a 8-year old greek boy with homozygous beta-thalassemia (author’s transl).]: Klin Padiatr, 1975; 187(4); 357-60 [in German]
24.. Dutta A, Dutta R, Sarda P, Jain J, Erythraemic myelosis in a thalassaemic child: J Indian Med Assoc, 1975; 65(8); 237-38
25.. Kurdi R, Rasjid A, Wahidiyat I, Thalassemia terminating in erythremic myelosis: Paediatr Indones, 1977; 17(3–4); 115-23
26.. Samal G, Sickle cell anemia with acute myeloid leukemia – (a case report): Indian Pediatr, 1979; 16(5); 453-54
27.. Pastore G, Miniero R, Morgando M, [Homozygous beta-thalassaemia and acute lymphoblastic leukaemia (author’s transl).]: Pediatr Med Chir, 1981; 3(4); 281-82
28.. Sandström H, Wahlin A, Eriksson M, Intravascular haemolysis and increased prevalence of myeloma and monoclonal gammopathy in congenital dyserythropoietic anaemia, type III: Eur J Haematol, 1994; 52(1); 42-46
29.. Byrnes RK, Dhru R, Brady AM, Congenital dyserythropoietic anemia in treated Hodgkin’s disease: Hum Pathol, 1980; 11(5); 485-86
30.. Hattori Y, Harada K, [Hereditary spherocytosis associated with Bence Jones type multiple myeloma: A case report.]: Rinsho Ketsueki, 1988; 29(2); 254-57 [in Japanese]
31.. Takegoshi T, Nishino T, Tanino M, an autopsy case of hemochromatosisand hepatoma combined with hereditary spherocytosis: Jpn J Med, 1984; 23(1); 48-52
32.. Kies MS, Abbott OD, Rubin RN, Recurrence of hemolysis in hereditary spherocytosis: A case due to leukemic infiltration of an accessory spleen: Mil Med, 1981; 146(1); 55-57
33.. Dvorák K, [Association of familial elliptocytosis and plasmacytoma with micromolecular paraproteinemia.]: Vnitr Lek, 1970; 16(7); 707-12 [in Czech]
34.. Arisawa K, Morita S, Kojima H, [Hereditary spherocytosis associated with non-Hodgkin’s lymphoma in the spleen.]: Rinsho Ketsueki, 1994; 35(9); 871-75 [in Japanese]
35.. Ishida Y, Niino M, Matsuda H, Bandou S, [Hereditary spherocytosis presenting with acute lymphoblastic leukemia.]: Rinsho Ketsueki, 1987; 28(3); 402-7 [in Japanese]
36.. Dulmovits BM, Appiah-Kubi AO, Papoin J, Pomalidomide reverses γglobin silencing through the transcriptional reprogramming of adult hematopoietic progenitors: Blood, 2016; 127(11); 1481-92
37.. Rychak E, Mendy D, Shi T, Pomalidomide in combination with dexamethasone results in synergistic anti-tumour responses in pre-clinical models of lenalidomide-resistant multiple myeloma: Br J Haematol, 2016; 172(6); 889-901
38.. Siegel DS, Schiller GJ, Song KW, Pomalidomide plus low-dose dexamethasone in relapsed refractory multiple myeloma after lenalidomide treatment failure: Br J Haematol, 2020; 188(4); 501-10
Figures
In Press
12 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943244
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943275
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943411
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942864
Most Viewed Current Articles
07 Mar 2024 : Case report
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report 
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250