26 December 2021: Articles 
Parathyroid Carcinoma: A Rare Endocrine Malignancy
Rare disease
Nádia Mourinho Bala1BDEF*, José Maria Aragüés1E, Sílvia Guerra1E, Nuno Cordeiro Raposo1B, Cristina Valadas1EDOI: 10.12659/AJCR.934221
Am J Case Rep 2021; 22:e934221






Abstract
BACKGROUND: Parathyroid carcinoma (PC) is an extremely rare endocrine malignancy, with a reported increase in incidence in the past decade. PC generally presents in an indolent fashion, featuring nonspecific symptoms associated with hypercalcemia.
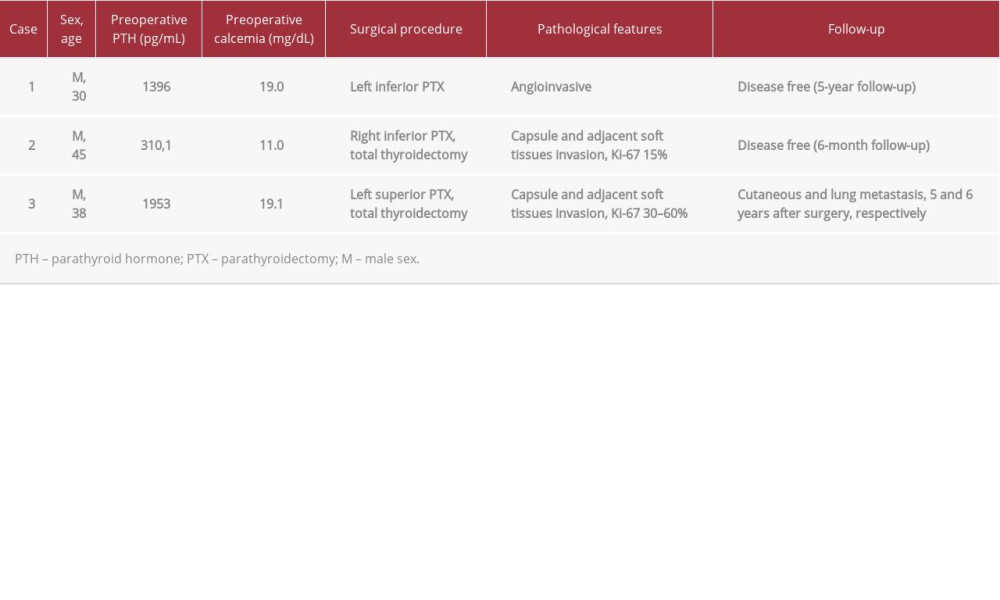
CASE REPORT: Case 1: A 30-year-old man was admitted for symptoms associated with hypercalcemia and elevated parathyroid hormone (PTH). Imaging examinations showed the presence of a cervical nodular lesion. The patient underwent surgery, and the pathological diagnosis was PC. Case 2: A 45-year-old man with a history of hypothyroidism was referred to our Endocrinology Department for a cervical nodular lesion. A fine-needle aspiration was performed, and the result was suggestive of papillary carcinoma. Blood testing showed only mild hypercalcemia and PTH elevation, with no associated symptoms. The patient underwent surgery, and the histological examination confirmed the diagnosis of PC. Case 3: A 38-year-old man presented with diffuse bone pain and muscle weakness, severe hypercalcemia, high levels of PTH, and a cervical mass. The patient underwent surgery. Diagnostic pathology confirmed the diagnosis of PC. Five years later, the patient presented with a cutaneous metastasis, followed 1 year later by pulmonary metastases.
CONCLUSIONS: Most PCs are slow-growing tumors. Some of these tumors are diagnosed in association with hereditary syndromes. A clear distinction between benign and malignant lesions is not always simple because there is a lack of specific clinical distinguishing features of malignant lesions. Currently, surgical resection is the preferred approach; however, owing to the rarity of this condition, there is a void of high-quality data.
Keywords: Hypercalcemia, Hyperparathyroidism, Parathyroid Cancer, Adult, Adult, Humans, Lung Neoplasms, Male, Parathyroid Hormone, Parathyroid Neoplasms
Background
Parathyroid carcinoma (PC) is the least common type of malignancy [1,2]. Owing to its extraordinary rarity, there is a scarcity of information regarding clinical and molecular factors associated with the etiology of this type of tumor [3]. PC represents less than 0.1% of all types of cancers and less than 1% of all causes of primary hyperparathyroidism [4–7]. PC can occur sporadically or as part of a genetic syndrome, most frequently as a part of hyperparathyroidism jaw tumor syndrome and less frequently, multiple endocrine neoplasia type 1 and type 2A [2,8]. Typically, PC behaves indolently, and most patients present with renal and bone-related symptoms [5,8]. No known clinically specific characteristics allow for the distinction between PC and parathyroid adenomas, although the severity of hypercalcemic symptoms tends to be more pronounced in PC [1–5,9]. There is a lack of randomized studies regarding the approach and prognosis of PC owing to the rarity of this condition, and to date, surgical therapy remains the mainstay approach. We share our experience by reporting 3 cases of PC.
Case Reports
CASE 1:
We report a case of a 30-year-old man with a past history of nephrolithiasis, which started approximately 10 years before, and an unremarkable family history. The patient was hospitalized with concerns of polydipsia, nausea, and muscle weakness for the previous 2 months. At the initial observation, the patient presented with a palpable and painless cervical nodule, which was approximately 40×30 mm and hard in consistency, with no other remarkable physical findings. Upon admission, the patient’s serum creatinine level was 1.83 mg/dL, despite no preexisting kidney disease. The remaining blood tests at this point revealed severe hypercalcemia of 19.0 mg/dL (reference range, 8.6–10.0 mg/dL), mild hypophosphatemia of 2.4 mg/dL (range, 2.5–4.5 mg/dL), and parathyroid hormone (PTH) of 1396 pg/mL (range, 15–65 pg/mL). The thyroid function was normal, and a cervical ultrasound was ordered. The ultrasound examination revealed the presence of a highly vascular, regular mass, 40 mm in the long axis, and adjacent to the left inferior thyroid pole. The high levels of PTH and severe hypercalcemia were indicative of primary hyperparathyroidism. Thus, a technetium-99m-sestamibi scintigraphy was performed, and there was an increased uptake of the radionuclide adjacent at the left inferior thyroid lobe. There were no signs of metabolic bone disease, namely osteitis fibrosa cystica. The patient was started on pamidronate a few weeks before surgery to treat the hypercalcemia. The potential diagnosis of PC was raised at this point owing to abnormally high calcium and PTH levels. When presented with the available treatment options, the patient opted for the resection of the left inferior parathyroid gland. Following the removal of the gland, the intraoperative PTH level dropped by more than 50%. The histologic examination confirmed the diagnosis of PC, with a long axis of 40 mm, vascular invasion, and no tissue invasion. Despite the conservative approach, 9 months after surgery, the patient showed no evidence of disease.
CASE 2:
We report the case of a 45-year-old man with a personal history of arterial hypertension and hypothyroidism due to Hashimoto thyroiditis. The patient was taking levothyroxine and ramipril. The family history was unremarkable. The patient was referred to our Endocrinology Department for a 28-mm cystic nodule in the posterior region of right thyroid lobe, with no associated symptoms. At first observation, the patient presented with a palpable nodule, hard in consistency, and located at the right thyroid lobe. A fine-needle aspiration cytology was performed, and the result was suggestive of papillary carcinoma. The patient’s laboratory testing showed a high PTH level of 310.1 ng/mL (reference range, 14–72 ng/mL), serum calcium of 11.0 mg/dL (range, 8.6–10.2 mg/dL), thyroid-stimulating hormone of 3.72 (range, 0.55–4.78), free T4 of 14.2 mol/L (range, 11.5–22.7 mol/L) and anti-thyroid peroxidase antibodies >1300 UI/mL (range, <60.0 UI/mL). Owing to the elevated PTH and serum calcium, which were suggestive of primary hyperthyroidism, a Tc99m-sestamibi parathyroid scintigraphy was ordered. The examination did not show any areas of increased uptake, and at this point, there was no suspicion of PC. Nonetheless, considering the cytological examination suggestive of papillary carcinoma, a total thyroidectomy was the chosen procedure. The histological specimen examination revealed a lymphoplasmocitary inflammatory infiltrate, suggestive of Hashimoto thyroiditis. The histologic examination of the excised nodule supported the diagnosis of PC, with capsular invasion, invasion of adjacent tissues, and no vascular invasion. The Ki-67 marker was 15%. Five years after surgery, the patient showed no evidence of disease. This patient did not, at any point, present with symptoms of nephrolithiasis or metabolic bone disease.
CASE 3:
We report the case of a 38-year-old man with an unremarkable past medical history. The patient was referred to our Endocrinology Department for bone pain and muscle weakness of a 6-month duration. At first observation, a palpable cervical nodule was noted, located at the left thyroid nodule. The initial laboratory testing showed severe hypercalcemia of 19.1 mg/dL (range, 8.6–10.2 mg/dL), hyperphosphatemia of 5.9 mg/dL, PTH of 1953 pg/mL (range, 14–72 pg/mL), thyroid function within the reference range, and a serum creatinine level of 3.52 mg/dL. The patient’s symptom duration and blood laboratory findings were suggestive of chronic disease, namely severe kidney injury with associated hyperphosphatemia. A cervical ultrasound was performed, which revealed the presence of a heterogeneous lesion, predominantly hypoechoic, lobulated, 66×31 mm in size, and by the superior and lateral aspects of left thyroid lobe. At this point, a fine-needle aspiration was performed since the origin of the nodule was unclear and the hypothesis of PC had not been considered. The cytological examination showed abundant chief cells with perinu-clear oil vacuole nuclei and the absence of colloid. Moreover, thyroglobulin immunostaining was reported as negative, favoring the parathyroid origin. A Tc99m-sestamibi parathyroid scintigraphy and bone scintigraphy were performed, and the results revealed an increased uptake of this lesion and of the bone tissue, which was diffuse and particularly evident in the skull, facial bones, and long bones of the arms and legs. A renal ultrasonography was requested, which showed medullary nephrocalcinosis and nephrolithiasis. Due to a high index of suspicion of PC, the preferred approach was total thyroidectomy and left parathyroidectomy. One month after surgery, the PTH values were within the reference range. The pathologic diagnosis was PC, with necrotic areas, gross calcifications, capsular invasion, vascular invasion, and tissue invasion with thyroid and muscle infiltration. The Ki-67 was in the 30% to 60% range. Three years later, we observed a steady increase of the PTH (from 123 to 200.1 pg/mL) and calcium values (from 9.8 to 11.3 mg/dL). Five years after the surgery, the diagnosis of a left supraclavicular cutaneous metastasis was established. This lesion was excised. Six years after the initial diagnosis, an F-fluorodeoxyglucose positron emission tomography (PET)/computed tomography (CT) scan was requested owing to rising levels of serum calcium (14.8 mg/dL) and PTH (1168 pg/mL). This examination showed 10 pulmonary nodules, suggestive of pulmonary metastases. The decided approach was the surgical excision of these lesions. New pulmonary lesions were detected on a follow-up CT scan, consistent with metastatic disease. In the initial lung resection surgery, a large amount of tissue was excised, making the surgical approach no longer a viable option for this patient because of the associated risk of respiratory failure. As an alternative, stereotactic body radiation therapy is currently being considered to hinder the rapid growth of the metastatic foci.
Discussion
PC is a very rare endocrine malignancy, affecting men and women equally, and usually presenting in the fifth and sixth decades of life [1,3,5]. In our series, 2 patients were unusually young, significantly under the reported average age for PC. None of the patients presented with other tumors, namely pheochromocytoma, medullary thyroid carcinoma, pituitary adenoma, or pancreatic tumor suggesting multiple endocrine neoplasia type 1 or type 2A syndrome. Roughly 90% of PC are functional [1,5]. Thus, most patients present with signs and symptoms of primary hyperparathyroidism, such as fatigue, muscle weakness, weight loss, renal disease (nephrolithiasis, nephrocalcinosis, acute kidney lesion), and bone disease (osteoporosis, osteitis fibrosa cystica, osteofibrosis) [3,5]. To date, the etiology of this tumor is not fully understood. PC mostly occurs in a sporadic fashion, with no associated conditions; however, previous exposure to radiation and secondary and tertiary hyperparathyroidism are considered risk factors. In rare cases, these tumors can be associated with genetic syndromes, the most frequent of these being hyperparathyroidism jaw tumor syndrome [1]. Extremely high blood calcium levels, often above 14 mg/dL, and PTH values of 3 to 15 times the upper limit of the reference range, are not unusual. These extreme alterations contrast with those found in benign conditions, which normally present with milder findings [2,4]. In the present report, 2 patients presented with severe hypercalcemia and PTH values 20 times the upper limit of the reference range (Table 1). However, 1 of the patients presented with mild hypercalcemia and a PTH elevation of only 4 times the upper limit of the reference range. In this particular case, owing to the mild symptoms at presentation, there was no suspicion of malignancy prior to surgery. There are no pathognomonic findings associated with PC. Nonetheless, PC lesions typically tend to be hypoechoic and lobulated tumors or larger (>3 cm), highly vascular tumors with poorly defined borders [1,3]. Performing a 99mTc-sestamibi parathyroid scintigraphy might aid in locating the tumor. If the diagnosis of PC is considered, a fine-needle aspiration examination should not be performed since there is a risk of tumoral dissemination along the needle track [5]. Capsular and vascular invasion are the only histopathologic pathognomonic findings associated with PC [1]. Surgical excision remains the cornerstone of the therapeutic approach. Generally, en bloc resection of the PC and ipsilateral hemithyroidectomy is the recommended procedure. Nonetheless, few authors recommend routine central neck dissection in the absence of clinical signs of tumoral invasion [4]. In most cases, an en bloc resection is not performed since PC is not considered as a likely diagnosis prior to surgery [5]. The follow-up of functional carcinomas is based on routine serum calcium levels and PTH screenings. If a significant change in these values is noted, an imagining examination should be performed, such as ultrasound, CT scan, magnetic resonance imaging, 99Tcsestamibi scintigraphy, or PET scan [2]. The 5-year survival rate of PC is 80% to 90% [4]. Metastatic disease at presentation and large tumor size (>3 cm) are associated with worse cancer-specific survival rates [10]. Most recurrence cases are local, but 25% of all patients might present with metastatic disease, most frequently lung, hepatic, and bone metastasis. Surgery is recommended and is effective at reducing the PTH and calcium levels in cases of recurrence, leading to an improvement in symptom control and temporary biochemical normalization. Chemotherapy and radiotherapy have shown disappointing results in the treatment of PC.
Conclusions
PC is an extremely rare entity, with only approximately 1000 cases reported in the literature. We described 3 cases, each with different associated epidemiology, clinical, and laboratorial findings and very different outcomes. Classically, these indolent tumors present generally with symptoms associated with acute kidney injury and hypercalcemia. Although there are no specific characteristics associated with malignancy, severe hypercalcemia and extreme PTH values should raise the index of suspicion for PC. An appropriately high index of suspicion is particularly important since it might impact the surgical approach, which at the time of this writing is considered the only potentially curative approach.
References:
1.. Ferraro V, Sgaramella LI, Di Meo G, Current concepts in parathyroid carcinoma: A single centre experience: BMC Endocr Disord, 2019; 19(1); 46
2.. Dudney WC, Bodenner D, Stack BC, Parathyroid carcinoma: Otolaryngol Clin North Am, 2010; 43(2); 441-53
3.. Betea D, Potorac I, Beckers A, Parathyroid carcinoma: Challenges in diagnosis and treatment: Ann Endocrinol (Paris), 2015; 76(2); 169-77
4.. Medas F, Erdas E, Loi G, Controversies in the management of parathyroid carcinoma: A case series and review of the literature: Int J Surg, 2016; 28; S94-S98
5.. Goswamy J, Lei M, Simo R, Parathyroid carcinoma: Curr Opin Otolaryngol Head Neck Surg, 2016; 24(2); 155-62
6.. Cetani F, Pardi E, Marcocci C, Update on parathyroid carcinoma: J Endocrinol Invest, 2016; 39(6); 595-606
7.. Mittendorf EA, McHenry CR, Parathyroid carcinoma: J Surg Oncol, 2005; 89(3); 136-42
8.. Cetani F, Pardi E, Marcocci C, Parathyroid carcinoma: Front Horm Res, 2019; 51; 63-76
9.. DoCao C, Aubert S, Trinel C, Parathyroid carcinoma: Diagnostic criteria, classification, evaluation: Ann Endocrinol (Paris), 2015; 76(2); 165-68
10.. Lo WM, Good ML, Nilubol N, Perrier ND, Patel DT, Tumor size and presence of metastatic disease at diagnosis are associated with disease-specific survival in parathyroid carcinoma: Ann Surg Oncol, 2018; 25(9); 2535-40
In Press
05 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942032
06 Mar 2024 : Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.942937
12 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943244
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943275
Most Viewed Current Articles
07 Mar 2024 : Case report 
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250