05 January 2022: Articles 
Delayed Hemolytic Transfusion Reaction in Sickle Cell Disease: A Case Series
Unknown etiology, Challenging differential diagnosis, Diagnostic / therapeutic accidents, Unusual setting of medical care, Educational Purpose (only if useful for a systematic review or synthesis)
Abrar J. Alwaheed1ABCDEFG, Safi G. AlqatariDOI: 10.12659/AJCR.934681
Am J Case Rep 2022; 23:e934681






Abstract
BACKGROUND: Transfusion therapy has a well-established role in the management of several sickle cell disease (SCD)-related complications. Nevertheless, the benefits of transfusion must outweigh the possible risks, including iron overload, infections, and transfusion reactions. Alloimmunization is the underlying etiology of most delayed hemolytic transfusion reactions (DHTR). DHTR is often underestimated and underdiagnosed in sickle cell disease patients as it mimics a vaso-occlusive crisis in presentation. Alloimmunization to RBC antigens can be a serious complication of transfusion, which is of particular interest in individuals with SCD, as the occurrence rate is higher in this population. This complication represents a secondary immunological phenomenon that typically arises after the emergence of an alloantibody to which the patient had been previously sensitized to.
CASE REPORT: Here, we report 2 cases of delayed hemolytic transfusion reaction (DHTR) in which the patients showed evidence of alloimmunization from previous blood transfusions. The patients were managed with a variety of medications, including supportive treatments, utilization of immunosuppressive agents, and enhancement of erythropoiesis. Both patients had evidence of clinical and laboratory improvement following the management.
CONCLUSIONS: DHTR is considered one of the most deleterious complications of transfusion in SCD patients. The diagnosis and management of DHTR is very challenging, especially because it can present differently in this population. A high index of clinical suspicion is needed in addition to the laboratory criteria.
Keywords: Anemia, Sickle Cell, Antigen-Antibody Reactions, Transfusion reaction, Anemia, Hemolytic, Autoimmune, Blood Transfusion, Humans, Isoantibodies
Background
Delayed hemolytic transfusion reactions (DHTR) are hyperhemolysis of both autologous and transfused red blood cells by an alloantibody that is usually not detected prior to transfusion [1]. Individuals initially become alloimmunized to red blood cell (RBC) antigens from previous transfusions or pregnancies. The immunological memory produced during prior transfusions will facilitate the generation of alloantibodies, which subsequently cause destruction of the RBCs [1]. DHTR usually occurs 24 h to 21 days after transfusion [2]. The appearance of clinical signs occurs a median of 9 days after blood transfusion [3]. It commonly manifests as fever, pain, jaundice, dark urine, and an unexplained decrease in hemoglobin after trans-fusion compared to the pretransfusion level [2,4].
A positive direct anti-globulin test or a positive antibody screening test is the hallmark serologic finding. Antibodies directed against Rh (D, C, E, c, e) and Kidd (Jka, Jkb) K, FY Ss system antigens are the antibodies most commonly implicated in DHTR. However, numerous other non-specific antibodies have been described [5,6]. Once alloantibodies are identified, the patient should receive antigen-negative RBCs to prevent future reactions. Of note, no antibodies are detected in at least 30% of the cases, which makes the diagnosis more difficult [7].
In general, DHTR is underdiagnosed because the clinical presentation is similar to vaso-occlusive crises (VOC) in sickle cell disease, which is far more prevalent than DHTR. In addition, patients with sickle cell disease and G6PD deficiency need long-term hemolysis and can present with acute hemolytic crisis requiring blood transfusions, which puts them at increased risk for transfusion-related complications, especially DHTR. As mentioned before, the post-transfusion screening tests tend to reveal false-negative results, making the diagnosis more challenging for physicians. This disease can be life-threatening in severe cases, and it usually requires immediate action [8].
According to the current literature, predicting the possible impact of various immunosuppressive medications when attempting to prevent or treat DHTRs is difficult. Future research is needed to determine the best therapeutic choices for DHTRs in individuals with sickle cell disease (SCD) and whether they have unique alloantibodies compared to the general population [9].
Case Reports
CASE 1:
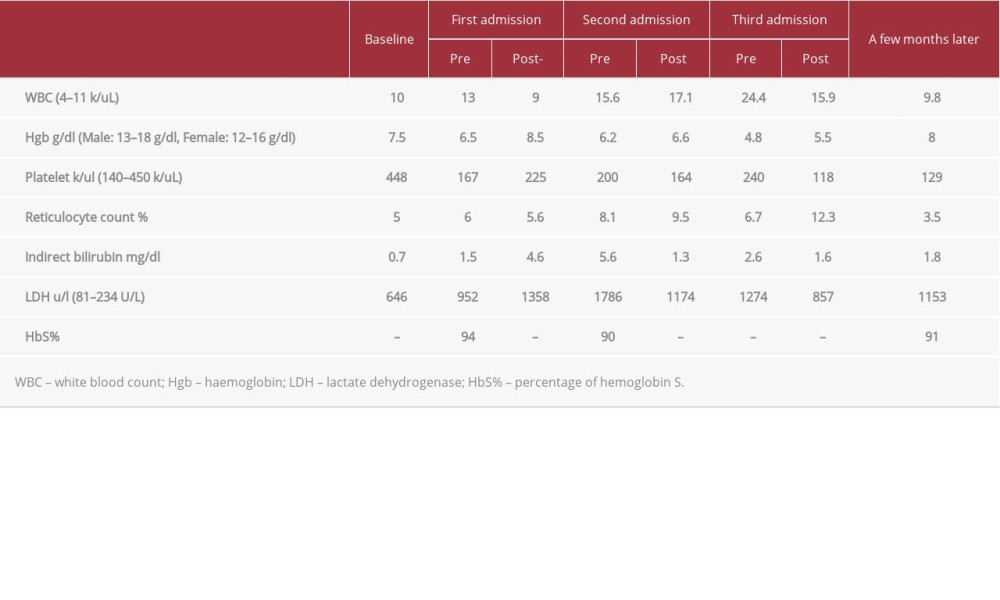
This was a 44-year-old Saudi woman known to have SCD and G6PD deficiency on folic acid replacement, with no known complications, and a baseline hemoglobin (Hgb) of 7.5–9 g/dl. She presented to the hospital reporting palpitations, shortness of breath, and fatigue for 4 months, which became worse 2 days prior to presentation. She was vitally and clinically stable, and a physical examination was unremarkable except for evidence of pallor. Her laboratory values showed a drop in hemoglobin to 6.5 g/dl, high LDH with a value of 952 U/L, leucocytosis with WBC of 13 k/uL, Hgb S saturation 94%, and her reticulocyte count was 6%. Table 1 shows the remainder of her laboratory values. Blood grouping and cross-matching were done. She had O+ blood group with negative DAT and antibodies screen. She received 1 unit of packed RBCs. Upon discharge, she was stable, and her Hgb improved to 8.5 g/dl. Table 1 shows the rest of her laboratory values.
Ten days later, she presented again with symptoms of anemia and high-grade fever of 39.5°C associated with chills. Physical examination showed evidence of pallor and jaundice. Her Hgb dropped again from 8.5 g/dl to 6.2 g/dl, WBC increased from 9 k/ul to 15.6 k/ul, LDH increased from 1358 U/L to 1786 U/L, indirect bilirubin increased from 4.6 mg/dl to 5.6 mg/dl, and her reticulocyte count was 8.1%. Table 1 shows the other laboratory values. DAT was positive and antibody screen was positive for anti-JK a antibody. Hemoglobin electrophoresis showed Hgb A 3.5% and Hgb S 90%. During her hospital stay, her Hgb dropped from 6.5 g/dl to 5.4 g/dl, for which she received 1 unit of packed RBCs and 2 doses of hydrocortisone 100 mg i.v. Upon discharge, her hemoglobin improved to 6.6 g/dl, WBC increased to 17.1 k/ul, and her LDH was 1174 u/l. Table 1 shows the other laboratory values upon discharge.
She presented again 4 days later with a high-grade fever reaching 39°C associated with increase leucocytosis with a WBC value of 24.4 k/ul. Her Hgb dropped from 6.6 g/dl to 4.8 g/dl. Table 1 shows the other laboratory values. The septic work-up was negative. She was diagnosed with alloimmunization and DHTR. Accordingly, she was managed with 2 doses of IVIG (0.5 g/kg then 1g/kg). She was also given darbepoetin alfa 60 mcg SQ twice per week for a total of 5 doses in the hospital, along with 3 doses of iron sucrose 200 mg as her ferritin level was 141.75 ng/ml. The patient also received 100-mg i.v. hydrocortisone injections 3 times per day for 3 days. The patient was eventually discharged with a Hgb value of 5.5 g/dl. Indirect bilirubin decreased to a value of 1.6 mg/dl, LDH decreased to a value of 857 U/L, and her reticulocyte count was 12.3% after being clinically stable with instructions to take darbepoetin 60 mcg SQ twice weekly for 6 weeks and to have regular clinic follow-up. She was evaluated in the clinic several times after that, with a stable Hgb of 7.5–8 g/dl and a reticulocyte count of 3.5%.
CASE 2:
A 35-year-old Saudi woman had a history of pernicious anemia and SCD diagnosed after her second delivery 7 years ago. She was in her usual state of health, with a baseline Hgb between 8–9 g/dl until 2 months before, when she was diagnosed with a chest infection, for which she received an antibiotic and was also transfused with 2 units of PRBC due to a low Hgb level.
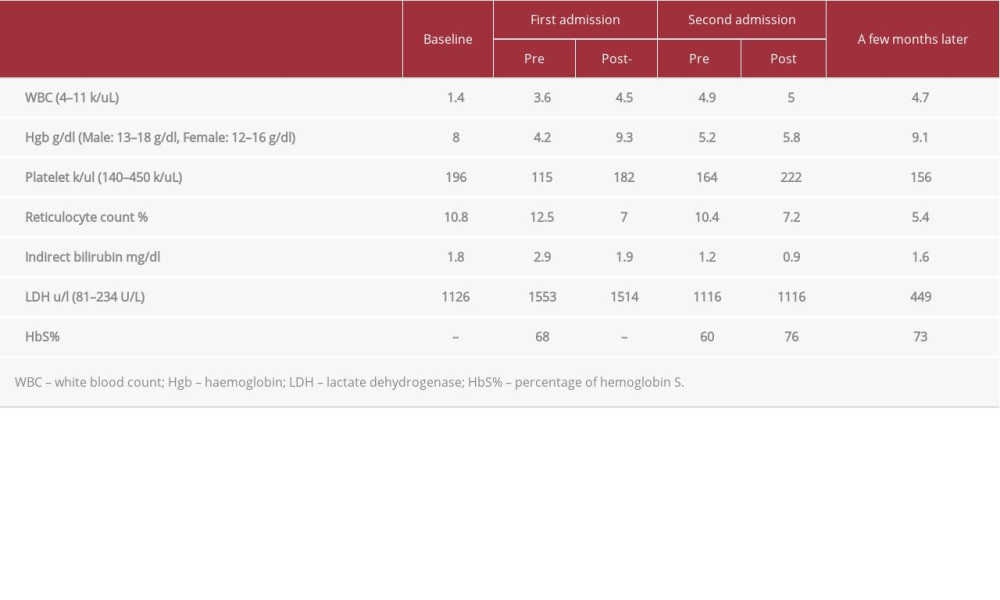
She presented to the hospital 2 weeks later reporting dizziness for 1 week, associated with palpitation and fatigue. She was vitally stable, with an unremarkable physical examination apart from pallor. Her Hgb level was 4.2 g/dl, indirect bilirubin level was 2.9 mg/dl, LDH level of 1153 U/L, and her reticulocyte count was 12.5%. The remainder of the laboratory values are shown in Table 2. Blood grouping and cross-matching were done. Her blood group was O+ and her DAT was positive. An antibodies screen was positive for Anti-C, Anti-E, Anti-K, and other non-specific antibodies. Hgb electrophoresis showed a Hgb A value of 7.4% and Hgb S of 68%. She was diagnosed as having alloimmunization and DHTR. The septic workup was negative. During her hospital stay she received a total of 4 units of PRBCs under supervision. Her post-transfusion Hgb increased to 9.3 g/dl and her reticulocyte count decreased from 12.5% to 7%. She also received 2 doses of 60 mg i.v. methylprednisolone and was discharged uneventfully 3 days later. Other laboratory values are shown in Table 2.
She presented again to the emergency room 6 days later reporting a fever reaching 38°C. Her Hgb dropped from 9.3 g/dl to 5.2 g/dl and the reticulocyte count increased from 7% to 10.4%. Other laboratory values are shown in Table 2. During the hospital course she received 2 doses of darbepoetin 60 mcg SQ to improve her HB, with avoidance of blood transfusion. She also received 1 dose of 100 mg hydrocortisone injection. Her hemoglobin upon discharge improved to 5.8 g/dl and the reticulocyte count decreased to 7.2%. The other laboratory values before discharge are shown in Table 2. She was discharged with instructions to continue vitamin B12 replacement and to follow up regularly in the hematology clinic. She was evaluated in the clinic a few months later with a stable Hgb between 7.5–8 g/dl and a reticulocyte count of 5.4%.
Discussion
A limited number of studies have investigated the incidence of DHTR, so its true incidence remains unknown. However, the prevalence of alloimmunization in SCD patients is reported to be between 30% and 50% and it is speculated to have increased recently [10]. The Incidence of DHTR in SCD has a very wide spectrum of 0.001–13.52% [1,4,10–12], making it the most common adverse event following occasional transfusion in patients with SCD [1]. The mortality rate in SCD patients is 6%–11%, suggesting that these reactions are not only more common than previously proposed, but are also likely to significantly affect SCD mortality rates [1].
Numerous studies have reported the risk factors for DHTR, which include a history of previous DHTR, and alloimmunization with antibodies [5]. In addition, having fewer previous transfusions was shown to be associated with a higher risk of developing DHTR. This can be explained by the fact that patients undergoing regular chronic transfusions often undergo extended RBC-matching compared to patients receiving episodic transfusions for acute indications [5].
Anoosha et al investigated the clinical features in addition to the outcomes of 99 DHTRs occurring in 69 referral centers. The most frequent DHTR-related clinical sign was dark urine/hemoglobinuria (94%). Most patients (89%) had a painful vasoocclusive crisis and 50% developed a secondary acute chest syndrome (ACS). Among these DHTRs, 61% developed in previously immunized patients and 28% developed in patients with prior DHTR [3]. In SCD, the clinical presentation of DHTR may be quite similar to acute painful crisis, with or without symptoms of aplastic crisis [13]. The anemia and pain are usually attributed to painful crises and hyperhemolysis; therefore, the diagnosis of DHTR may be delayed [8]. In addition to the clinical manifestation of acute or delayed hemolysis, these patients often experience painful crises, may show evidence of reticulocytopenia, and can develop more severe anemia than before the transfusion [13].
Racial differences in RBC antigen expression between recipients (predominantly African Americans) and donors (predominantly Whites) are thought to contribute to the greater likelihood of alloimmunization in SCD patients. A study comparing the red cell morphologies of African American patients and blood bank donors discovered that donors had a much higher occurrence of the antigens E, C, Kell, Fya, Fyb, and Jkb, which could contribute to the higher incidence of alloimmunization in this population [13]. Both patients in this case report had documented evidence of alloimmunization. The first patient had +ve Anti-Jka antibodies and the second patient had +ve Anti-C, Anti-E, Anti-K, and other non-specific antibodies [13].
According to the American Society of Hematology (ASH), DHTR is defined as a drop in hemoglobin within 21 days following transfusion, with 1 or more additional criteria. These criteria include development of new RBC alloantibody, haemoglobinuria, increase in hemoglobin S level and decrease in hemoglobin A level after transfusion, which were present in our patients. Development of reticulocytosis or relative reticulocytopenia in comparison to the baseline for the patients can also occur in DHTR with a marked increase in LDH [7]. The patients in this case report had variations in their reticulocyte counts and other markers of haemolysis like LDH and indirect bilirubin at every admission.
In the second case, the reticulocyte count improved after management in each admission and eventually returned to baseline within a few months. However, in the first case the reticulocyte counts kept fluctuating in an independent manner without correlation with the management in each admission. Therefore, diagnosis based on markers of hemolysis alone can be challenging, especially in SCD patients, who commonly have ongoing chronic hemolysis.
There are no validated treatment protocols for DHTR and no consensus on the standard dosage for the suggested treatments due to the lack of well-designed randomized-controlled trials. The general treatment recommendations for DHTR include avoidance of further transfusion, use of immunosuppressive therapy, and optimizing erythropoiesis. In addition to supportive management, several treatment modalities are commonly utilized, including IVIG for prevention of antibody-mediated immune destruction and corticosteroids in high doses for immune system modulation [14]. However, due to the absence of a unified dosing system, each hospital uses a different protocol for dosing and duration of the following medications. For example, a study by Kate et al published in the British Journal of Hematology (BJH) recommended using IVIG in a dose of 1 g/kg/d for 1–2 days [2], whereas Sawsan et al recommended using it in a dose of 0.4 g/kg/day for 5 days [15]. Steroids have been found to act in a synergistic manner with the IVIG to supress macrophage activity [14]. A report by Kate et al, published in th British Journal of Hematology, implemented a protocol administering steroids in the form of methylprednisolone in a dose of 0.5 g daily for up to 5 days [2], whereas Sawsan et al utilized it in a dose of 1 mg/kg daily in the form of prednisolone tablets for 1 week [15]. In addition to the previous medications, recombinant EPO and IV iron have been reported to boost endogenous RBC production in the setting of severe anemia [1]. Since ferritin level is considered an acute-phase reactant, which is expected to be elevated in most patients with SCD, this makes it difficult to find a standard definition for iron deficiency in these patients. However, Gardner et al suggested transferrin saturation of less than 20% can be used as indication for i.v. iron [2]. High doses of EPO, at 250–800 u/kg/dose 3 times weekly, were recommended by Kate et al [2], whereas France et al and others from the American Society of Hematology recommend darbepoetin at a dose of 100–300 g every 48 h [7,14,16]. Some studies described the use of rituximab for patients with prior DHTR who had evidence of alloimmunization to prevent further alloimmunization [3,6]. Kate et al proposed using rituximab in both low doses 100 mg i.v. once per week for 4 weeks or in standard doses of 375 mg/m2 weekly for 4 weeks [2]. In contrast, Clarisse et al utilized rituximab in a dose of 1000 mg i.v. as a single dose [17]. Recent evidence reported by Swee et al suggests that excessive complement activation may account for the most severe DHTR presentations with accompanying hyperhemolysis [3,6]. Therefore, eculizumab can be one of the treatment options in patients experiencing DHTR-associated hyperhemolysis to prevent irreversible multi-organ failure and it results in rapid clinical improvement [1]. France et al suggested using eculizumab with a dose of 900 mg on day 1 and day 7 only in the presence of severity criteria, which consists of ACS with hypoxemia or acute pulmonary HTN, stroke, and organ failure [14,17].
Although additional studies are needed, these reports provide us with more options for effective treatment, especially for severe cases [1,14].
DHTR events may be considered as a prompt to initiate hydroxyurea in patients who are considered un-transfusable to maximize their HB levels and minimize transfusion needs in the longer term [18]. Ultimately, the most important measure is to withhold further transfusions to avoid accelerated haemolysis. However, in certain cases with profound anemia and worsening hypoxemia or heart failure, the administration of additional transfusions may become unavoidable as a measure of last resort [11].
In comparison to these protocols, the patients discussed in our case series were considered to have mild to moderate DHTR and thus were not candidates for immunosuppression by eculizumab or rituximab. IVIG and steroids were provided in both cases, as they are the first-line medications in most of the recommendations/studies. Supportive management is the cornerstone for treating DHTR and it should focus on minimizing RBC transfusion unless in life-threatening anemia, enhancing erythropoiesis, and replacing the common hematopoietic deficiencies by administering iron, folic acid, vitamin B12, and EPO, which were given to our patients.
Some of the obstacles we encountered during the management of these cases were the absence of a clear dosing system, the type of steroid used, and the limited resources available, since some of these medications have a high cost. This could be overcome by creating a unified hospital DHTR treatment protocol, which we are developing now. Unfortunately, both patients mentioned in our case series declined hydroxyurea use, even though it could benefit them in the long term, as suggested previously.
Conclusions
DHTR is considered one of the most deleterious complications of transfusion in SCD patients. The diagnosis and management of DHTR is very challenging, especially because it can present differently in this population. A high index of clinical suspicion is needed in addition to the laboratory criteria. Further research and clinical trials are needed in this field to explore additional treatment options such as the utilization of immunosuppressants and supportive medications with their optimal doses, while considering the potential adverse effects, cost, and the rate of improvement in patient outcomes.
References:
1.. Thein SL, Pirenne F, Fasano RM, Hemolytic transfusion reactions in sickle cell disease: Underappreciated and potentially fatal: Haematologica, 2020; 105(3); 539-44
2.. Gardner K, Hoppe C, Mijovic A, Thein SL, How we treat delayed haemolytic transfusion reactions in patients with sickle cell disease: Br J Haematol, 2015; 170(6); 745-56
3.. Habibi A, Mekontso-Dessap A, Guillaud C, Delayed hemolytic transfusion reaction in adult sickle-cell disease: Presentations, outcomes, and treatments of 99 referral center episodes: Am J Hematol, 2016; 91(10); 989-94
4.. Coleman S, Westhoff CM, Friedman DF, Chou ST, Alloimmunization in patients with sickle cell disease and underrecognition of accompanying delayed hemolytic transfusion reactions: Transfusion, 2019; 59(7); 2282-91
5.. Narbey D, Habibi A, Chadebech P, Incidence and predictive score for delayed hemolytic transfusion reaction in adult patients with sickle cell disease: Am J Hematol, 2017; 92(12); 1340-48
6.. Guo A: Managing alloimmunization, delayed hemolytic transfusion reactions, and hyper-hemolysis in sickle cell disease [Internet], 2020, Boston University Libraries OpenBU [cited 2021Nov17]. https://openbu.edu/handle/2144/39420
7.. Chou ST, Alsawas M, Fasano RM, American Society of Hematology 2020 guidelines for sickle cell disease: Transfusion support: Blood Adv, 2020; 4(2); 327-55
8.. Zerra P, Josephson C, Delayed hemolytic transfusion reactions: Transfusion Medicine and Hemostasis, 2019; 397-400
9.. Dean CL, Maier CL, Chonat S, Challenges in the treatment and prevention of delayed hemolytic transfusion reactions with hyperhemolysis in sickle cell disease patients: Transfusion, 2019; 59(5); 1698-705
10.. Buetens O, Shirey RS, Goble-Lee M, Prevalence of HLA antibodies in transfused patients with and without red cell antibodies: Transfusion, 2006; 46(5); 754-56
11.. Mpinganzima C, Haaland A, Holm AG, Two consecutive episodes of severe delayed hemolytic transfusion reaction in a sickle cell disease patient: Case Rep Hematol, 2020; 2020; 2765012
12.. Kanti Sinha RT, Rai P, Dey A, A study of transfusion related adverse events at a tertiary care center in Central India: A retrospective evaluation: Journal of Medical Sciences and Health, 2016; 2(3); 6-12
13.. Scheunemann L, Ataga K, Delayed hemolytic transfusion reaction in sickle cell disease: Am J Med Sci, 2010; 339(3); 266-69
14.. Pirenne F, Yazdanbakhsh K, How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions: Blood, 2018; 131(25); 2773-81
15.. Omer SA, Alaesh JS, Algadeeb KB, delayed hemolytic transfusion reaction in a patient with sickle cell disease: Case report: Int Med Case Rep J, 2020; 13; 307-11
16.. Saleh M, Mallipeddi VP, Ali A, Delayed hemolytic transfusion reaction in a sickle cell disease patient: A case report: Cureus, 2020; 12(12); e12167
17.. Radia D, Momoh I, Dillon R, Anemia management: development of a rapidaccess anemia and intravenous iron service: Risk Manag Healthc Policy, 2013; 6; 13-22
18.. Sahu S, Hemlata , Verma A, Adverse events related to blood transfusion: Indian J Anaesth, 2014; 58(5); 543-51
In Press
06 Mar 2024 : Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.942937
12 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943244
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943275
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943411
Most Viewed Current Articles
07 Mar 2024 : Case report 
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250