30 August 2022: Articles 
Thrombotic Thrombocytopenic Purpura Triggered by Acute Myeloid Leukemia: A Case Report
Rare coexistence of disease or pathology
Ijele Adimora1E*, Cristhiam M. Rojas HernandezDOI: 10.12659/AJCR.935911
Am J Case Rep 2022; 23:e935911






Abstract
BACKGROUND: Acute myeloid leukemia (AML) is a myeloid progenitor malignancy characterized by clonal expansion of immature blasts. Complications of AML can result from disease-related or treatment-related complications and commonly include bleeding and disseminated intravascular coagulation. Thrombotic thrombocytopenic purpura (TTP) is a microangiopathy syndrome characterized by a mechanical hemolytic anemia and a consumptive thrombocytopenia resulting in end-organ damage from thrombotic occlusion of small vessels.
CASE REPORT: We describe a case of TTP at our institution that developed after diagnosis of AML, an exceedingly rare phenomenon with only one such documented case in the current literature. We were advised to see this patient after development of renal failure and encephalopathy. Suspicion for TTP was initially low, as our patient had a low pre-test probability of TTP by the PLASMIC score. Our patient was treated for disseminated intravascular coagulopathy, without response. Plasma exchange pheresis (PLEX) was eventually begun 3 days after presentation upon result of ADAMTS13 activity at 10%, with presence of inhibitor. ADAMTS13 activity levels were used to guide continuation of PLEX, given our patient’s persistent pancytopenia.
CONCLUSIONS: Our case demonstrates the challenges of identifying and managing TTP in patients with concomitant hematologic malignancies. ADAMTS13 activity levels should be collected in patients presenting with evidence of hemolytic anemia, even if the pre-test probability of TTP is low.
Keywords: Anemia, Hemolytic, Leukemia, Myeloid, Acute, Purpura, Thrombotic Thrombocytopenic, Disseminated Intravascular Coagulation, Humans, Plasma Exchange
Background
Acute myeloid leukemia (AML) is a myeloid progenitor malignancy characterized by clonal expansion of abnormal blasts. Proliferation of abnormal blasts leads to a disruption of normal hematopoiesis in the bone marrow, leading to bone marrow failure. Complications of AML can result from disease-related or treatment-related complications. Bleeding is a common complication that can result from thrombocytopenia, disease infiltration to the liver, or disseminated intravascular coagulation [1–3].
Thrombotic thrombocytopenic purpura (TTP) is a microangiopathy syndrome characterized by a mechanical hemolytic anemia and a consumptive thrombocytopenia resulting in end-organ damage from thrombotic occlusion of small vessels. TTP can be either congenital or acquired, with the acquired form being most common. The acquired form is autoimmune, caused by the development of antibodies to ADAMTS13, a proteolytic enzyme which cleaves von Willebrand factor [4]. The most commonly identified triggers for acquired TTP are bacterial and viral infections, autoimmune diseases, pregnancy, and drugs. The occurrence of thrombotic microangiopathy (TMA) is not uncommon in patients with metastatic cancer and can also be incited by chemotherapeutic agents; however, TTP should be viewed as a microangiopathic syndrome that is distinctly associated with severe ADAMTS13 deficiency. ADAMTS13 deficiency leads to uncleaved von Willebrand factor multimers and dysregulated platelet adhesion. TTP, left untreated, has a mortality rate of approximating 90%, and therefore patients with suspected TTP are treated with plasma exchange (PLEX) prior to resulting ADAMTS13 levels [5,6].
Case Report
The patient was a 71-year-old man with a past medical history of benign prostatic hyperplasia, hypertension, hyperlipidemia, and stage III chronic kidney disease and no prior history of hematologic malignancy who presented to an outside institution with a 3-week history of fatigue, fevers, chills, and dyspnea. At the outside institution, he was found to have a hemoglobin level of 6.8 g/dL, platelet count of 18 000/µL, and white blood cell count of 27.2×103/µL, with 33% peripheral blasts. Computed tomography of the chest revealed hilar and mediastinal lymphadenopathy. He was then transferred to our institution for concern of acute leukemia.
At presentation to our institution, he was in stable condition but febrile. The remainder of his examination was normal. His white blood cell count had increased to 33.6×103/µL, with 58% circulating blasts. His creatinine level was near his baseline, at 1.49 mg/dL.
The patient was started on hydroxyurea, which was followed by the 1-day administration of cytarabine for urgent cytoreduction, given the patient’s high blast count. Blood cultures were obtained and were negative. Cefepime, caspofungin, and valacyclovir were begun empirically.
A bone marrow biopsy with aspirate was obtained, revealing 78% myeloblasts by flow cytometry on aspirate. Routine cytogenetic testing revealed aneuploidy with complex karyo-type: 44,XY, del(5)(q11.2q33),del(6)(q13q23), add(7)(p13), add(9)(q13), der(17)t(17;20)(q11.2;q11.2)del(20)(q11.2q13.3), -18,add(18)(p11.2), -19, der(20)t(17;20). Next generation sequencing detected a sole TP53 mutation with no other somatic mutations identified. Pathological analysis confirmed that these findings were consistent with acute myeloid leukemia (de novo AML, AML not otherwise specified (NOS) by WHO classification).
On hospital day 3, the patient developed encephalopathy and tachypnea, with worsening kidney injury and metabolic acidosis. He was transferred to the Intensive Care Unit to begin continuous renal replacement therapy, which was continued for 3 days. He was started on decitabine and venetoclax for treatment of AML, given his high-risk cytogenetics and multiple complications. By hospital day 8, the patient’s creatinine level had risen to 5.4 mg/dL, and he was started on continuous veno-venous hemodialysis.
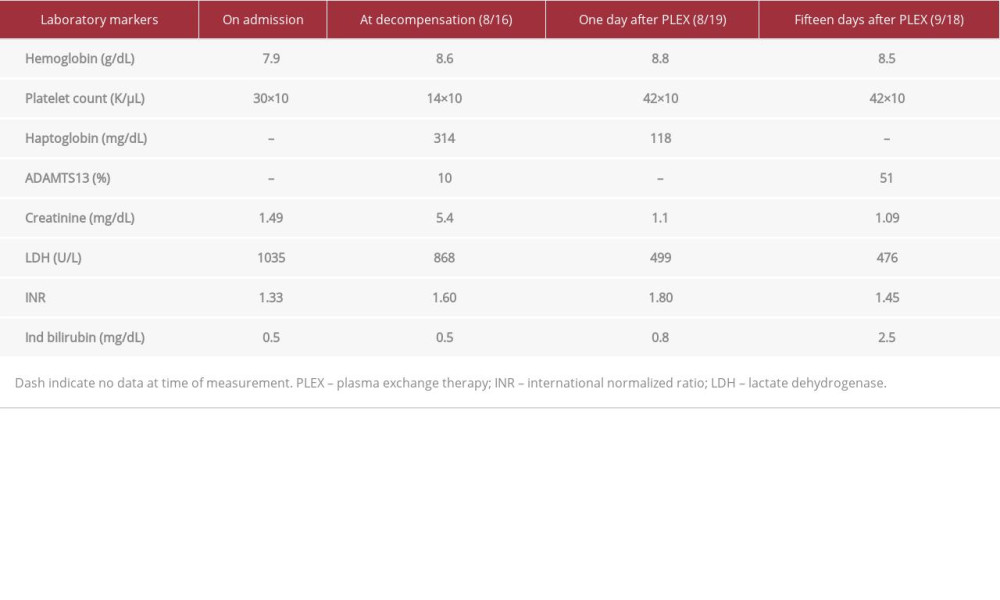
On hospital day 9, the Hematology Department was consulted for evaluation of the patient’s worsening acute kidney injury and pancytopenia. A peripheral smear showed 3 to 4 schistocytes per high-power field (Figure 1). Lactate dehydrogenase (LDH) and haptoglobin levels were elevated (Table 1). A direct Coombs test was performed and was negative for both IgG and C3. The international normalized ratio was also elevated at 1.62, and the patient had developed a right upper extremity deep vein thrombosis with daily extension of clot burden. Given these values and the patient’s clinical presentation, our differential diagnosis at the time primarily included cancer-associated TMA and subclinical disseminated intravascular coagulopathy. The patient was treated with supportive measures; however, he remained unresponsive to multiple platelet transfusions and cryoprecipitate. ADAMTS13 activity level and
The patient received a total of 1 cycle of decitabine and venetoclax combined regimen. Over his subsequent hospital course, his mental status worsened. Repeat blood cultures revealed
Discussion
The natural clinical course of TTP varies widely, but TTP should be suspected in all patients who present with microangiopathic hemolytic anemia (elevated LDH, decreased haptoglobin, and evidence of schistocytes on smear) and thrombocytopenia. Additional symptoms of fever, altered mental status, and kidney injury are present in only 7% of patients by some estimates [7].
An ADAMTS13 activity level less than 10% is highly specific for TTP [8]. However, at many institutions it can take several days to weeks to result. Because a delay in initiation of PLEX therapy could result in higher rates of mortality, there are several clinical prediction tools which help gauge the pre-test probability of TTP diagnosis. These tools can help guide clinicians in initiation of urgent PLEX. One such tool, as mentioned, is the PLASMIC score. The PLASMIC score helps stratify patients into low, intermediate, or high risk of TTP [9]. The score takes into account kidney function, evidence of hemolysis and platelet count, and other factors [9,10]. Although a low PLASMIC score did not prompt the empiric initiation of PLEX in our patient, fortunately an ADAMTS13 level was still obtained.
Disseminated intravascular coagulopathy and TMA are known to present in patients with active malignancy. TMA is a term that describes microvascular abnormalities leading to micro-vascular thrombosis, which may or may not present with micro-angiopathic hemolytic anemia. The spectrum of TMA includes syndromes such as TTP, Shiga toxin-mediated hemolytic uremic syndrome, complement-mediated TMA (commonly referred to as atypical hemolytic uremic syndrome), and drug-induced TMA (DITMA). Although TTP is a syndrome under the umbrella of TMA, it is distinct from all other syndromes included under the TMA nomenclature because it is defined by acquired or inherited ADAMTS13 deficiency. Autoantibodies to ADAMTS13 are not directly associated with hematologic malignancies; however, microvascular thrombi and microangiopathies can develop through other pathways [11].
Both macrovascular and microvascular thromboses routinely occur in malignancy. Mucinous-producing tumors of the gastrointestinal tract, pancreas, and ovaries are associated with venous thromboembolism, and hematologic malignancies, such as acute promyelocytic leukemia, can induce disseminated intravascular coagulopathy [12]. Widespread activation of the complement system is another mechanism of TMA thought to occur in malignancy. Metastatic cancer directly activates the complement system via tumor-induced upregulation of complement proteins, thereby resulting in complement-mediated hemolysis. Increased cancer-mediated thrombin generation can also lead to increased activation of C5a, resulting in increased platelet aggregation and microthrombi formation [11]. Finally, many patients with cancer commonly develop DITMA from cancer therapies [13]. Examining the different classes of therapeutic agents most known to trigger TMA (the anti-VEGF inhibitors bevacizumab, mitomycin, and gemcitabine, and platinum-based agents, such as cisplatin, among others), hydroxyurea has not been described to be among them [13]. There are reports of decitabine causing a thrombotic microangiopathy with primarily renal involvement [14]; however, our patient began to decompensate prior to having received decitabine therapy.
Studies have suggested that TTP may be over-diagnosed in patients presenting with microvascular hemolysis and ADAMTS13 activity levels >10%. This can lead to the inappropriate use of PLEX and the under-utilization of complement inhibitors, such as eculizumab [15]. While an ADAMTS13 activity level >10% alone cannot distinguish TTP from complement-mediated TMA, TTP should be strongly considered over complement-mediated TMA in patients with severe renal deficiency, severe thrombocytopenia, and an ADAMTS13 activity level <10% [15].
Here, we described a patient with acute myeloid leukemia who developed a severe acquired deficiency of ADAMTS13, an exceedingly rare phenomenon. To the best of our knowledge, this is among the first few cases reporting severe microangiopathy from TTP debuting in a patient with a diagnosis of AML [6]. In this case study, we described the challenges in the diagnosis, management, and treatment monitoring of TTP in the setting of AML.
Our patient’s diagnosis could not be distinguished by his clinical picture alone. His hypofibrinogenemia, renal failure, refractory thrombocytopenia, and deep vein thrombosis could be explained by complement-mediated TMA and disseminated intravascular coagulopathy. Obtaining the ADAMTS13 activity level and
After PLEX is begun in the treatment of TTP, daily PLEX is continued until a platelet count of 150×109 has been reached. However, because of the degree of myelosuppression from chemotherapy and active malignancy, platelets could not be used as a reliable marker to assess PLEX response. There is literature to suggest that ADAMTS13 activity monitoring can be an effective way to assess for TTP remission. A study of 57 patients by George et al found that the relative risk of relapse in patients who had ADAMTS13 activity of less than 15% was 4.01 (
Conclusions
Our case demonstrates the difficulty in diagnosing TTP in patients with hematologic malignancies. A high index of suspicion must be maintained in these patients, and PLASMIC score should be interpreted with caution. Early ADAMTS13 activity collection is paramount in making a timely diagnosis and should not be delayed. ADAMTS13 activity can be monitored for response to PLEX in patients with persistent thrombocytopenia; however, activity does not always correlate with TTP remission. More studies are needed to determine the utility of using ADAMTS13 activity to guide the frequency of PLEX administration in patients with hematologic malignancies. Finally, we describe one of very few cases of acquired TTP diagnosis in the setting of newly diagnosed AML. The trigger for our patient’s episode of TTP remains unclear, as severe ADAMTS13 deficiency does not commonly co-occur in patients with cancer-associated TMA, and the chemotherapeutic agent our patient received has not been described to trigger drug-induced TMA. More studies are needed to examine whether autoantibodies against ADAMTS13 can be incited by AML, and what role hydroxyurea and other chemotherapeutic agents may play in the development of TTP in patients with hematologic malignancies.
References:
1.. De Kouchkovsky I, Abdul-Hay M, Acute myeloid leukemia: A comprehensive review and 2016 update: Blood Cancer J, 2016; 6(7); 441
2.. Khwaja A, Bjorkholm M, Gale RE, Acute myeloid leukaemia: Nat Rev Dis Primers, 2016; 2; 16010
3.. Short NJ, Rytting ME, Cortes JE, Acute myeloid leukaemia: Lancet, 2018; 392(10147); 593-606
4.. Chiasakul T, Cuker A, Clinical and laboratory diagnosis of TTP: An integrated approach: Hematology, 2018; 1; 530-38
5.. Mancini I, Pontiggia S, Palla R, Artoni A, Clinical and laboratory features of patients with acquired thrombotic thrombocytopenic purpura: Fourteen years of the Milan TTP registry: Thromb Haemost, 2019; 119(5); 695-704
6.. Kucharik MP, Waldburg D, Chandran A, Acute myeloid leukemia presenting as thrombotic thrombocytopenic purpura: Cureus, 2020; 12(2); 6869
7.. Lotta LA, Mariani M, Consonni D, Different clinical severity of first episodes and recurrences of thrombotic thrombocytopenic purpura: Br J Haematol, 2010; 151(5); 488-94
8.. Kalish Y, Rottenstreich A, Rund D, Hochberg-Klein S, Atypical presentations of thrombotic thrombocytopenic purpura: A diagnostic role for ADAMTS13: J Thromb Thrombolysis, 2016; 42(2); 155-60
9.. Paydary K, Banwell E, Tong J, Diagnostic accuracy of the PLASMIC score in patients with suspected thrombotic thrombocytopenic purpura: A systematic review and meta-analysis: Transfusion, 2020; 60(9); 2047-57
10.. Tang N, Wang X, Li D, Sun Z, Validation of the PLASMIC score, a clinical prediction tool for thrombotic thrombocytopenic purpura diagnosis, in Chinese patients: Thromb Res, 2020; 172; 9-13
11.. Weitz IC, Thrombotic microangiopathy in cancer: Thromb Hemost, 2019; 45(04); 348-53
12.. Abdol Razak NB, Jones G, Bhandari M, Cancer-associated thrombosis: An overview of mechanisms, risk factors, and treatment: Cancers, 2018; 10(10); 380
13.. Qin AB, Tan Y, Su T, Decitabine-induced kidney thrombotic microangiopathy with glomerular crescents formation and tubular necrosis: A case report: Medicine, 2020; 99(43); e22901
14.. Al-Nouri ZL, Reese JA, Terrell DR, Drug-induced thrombotic microangiopathy: A systematic review of published reports: Blood, 2015; 125(4); 616-18
15.. Cataland SR, Yang S, Wu HM, The use of ADAMTS 13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies: Br J Haematol, 2012; 157(4); 501-3
16.. Page E, Kremer Hovinga A, Terrell R, Clinical importance of ADAMTS13 activity during remission in patients with acquired thrombotic thrombocytopenic purpura: Blood, 2016; 128(17); 2175-78
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133