29 November 2023: Articles ")
Co-Occurring Thrombotic Thrombocytopenic Purpura and Autoimmune Hemolytic Anemia in a Child Carrying the Pathogenic SHOC2 c.4A>G (p.Ser2Gly) Variant
Rare disease
Lijun Liu1BCDE, Chanchan Hu1BFG, Zhenjie Chen1ADG*, Shuzhen Zhu2BCD, Lvchang Zhu1CEFDOI: 10.12659/AJCR.942377
Am J Case Rep 2023; 24:e942377






Abstract
BACKGROUND: RASopathies involve mutations in genes that encode proteins participating in the RAS-mitogen-activated protein kinase pathway and are a collection of multisystem disorders that clinically overlap. Variants in the SHOC2 gene have been reported in Noonan-like syndrome, which include distinct facial features, short stature, congenital cardiac defects, developmental delays, bleeding disorders, and loose anagen hair. This report is of a 7-year-old girl with the c.4A>G (p.Ser2Gly) variant of the SHOC2 gene, consistent with Noonan-like syndrome, with loose anagen hair, presenting with thrombotic thrombocytopenic purpura and autoimmune hemolytic anemia.
CASE REPORT: The child had a medical history of 7 hospitalizations at our institution. At the age of 2 months, she underwent surgical correction for ventricular and atrial septal defects. At the age of 2 years, tonsil and adenoid removal surgery was performed, followed by surgery for otitis media at age 5 years. At 7 years, she was hospitalized for the simultaneous occurrence of thrombotic thrombocytopenic purpura and autoimmune hemolytic anemia. The patient displayed short stature and mild intellectual disability. Notable facial features included sparse hair, mild frontal bossing, and low-set ears. Antinuclear antibody levels demonstrated a significant gradual shift. Through trio whole-exome sequencing, a c.4A>G (p.Ser2Gly) variation in the SHOC2 gene was identified.
CONCLUSIONS: Given the clinical information and genetic testing results, the patient’s condition appeared to closely be a type of RASopathy. This report has highlighted the importance of physical, developmental, and genetic testing in children presenting with dysmorphism, developmental delay, and hematological abnormalities.
Keywords: Anemia, Hemolytic, Autoimmune, Thrombotic thrombocytopenic purpura, acquired, SHOC2 Protein, Human, Noonan-Like Syndrome with Loose Anagen Hair, Female, Humans, Child, Child, Preschool, Infant, Phenotype, Purpura, Thrombotic Thrombocytopenic, Noonan Syndrome, Mutation, Intracellular Signaling Peptides and Proteins
Background
RASopathies, which involve mutations in genes that encode proteins participating in the RAS-mitogen-activated protein kinase (RAS/MAPK) pathway, are a collection of multisystem disorders that clinically overlap and are among the most prevalent developmental disorders in humans [1]. The RAS pathway plays a crucial role in regulating the cell cycle, cellular growth, replication, differentiation, and metabolism [2]. It is essential for establishing and maintaining tissue homeostasis and balance in various tissues. The SHOC2 c.4A>G (p.Ser2Gly) variant is commonly seen in Noonan syndrome (NS; OMIM 163950) or Noonan-like syndrome with loose anagen hair (NS/ LAH; OMIM 607721) case reports [3–11]. NS/LAH is an auto-somal dominant RASopathy characterized by features that resemble NS [12–15]. Thrombotic thrombocytopenic purpura (TTP) is a prevalent form of microangiopathic hemolytic anemia (MAHA) characterized by the pathological development of hyaline thrombi in the extensive microcirculation [16,17]. Meanwhile, autoimmune hemolytic anemia (AIHA) is a condition characterized by the presence of autoantibodies in the bloodstream [18,19]. The coexistence of both MAHA and AIHA in a single patient is a rare phenomenon. The present article represents a single-case, 10-year retrospective study. A retrospective investigation was conducted involving a 7-year-old girl exhibiting the uncommon coexistence of TTP and AIHA. The clinical data, along with genetic analysis, strongly indicate that the child was affected by a form of RASopathy attributed to the SHOC2 gene and concurrently experienced the development of immune dysregulation.
Case Report
The present study pertains to the case of a pediatric patient with the co-occurrence of TTP and AIHA. The research involved the analysis of data from 7 hospital admissions at our institution spanning a 10-year period from December 2012 to December 2022 (Table 1). All clinical information was extracted from the patient’s medical records, and descriptive analyses were conducted on all variables. The present study was approved by the Ethics Committee of the Children’s Hospital, Zhejiang University School of Medicine.
The child had a medical history of 7 hospitalizations at our institution. Notably, at the age of 2 months, she underwent surgical correction for ventricular and atrial septal defects. At the age of 2 years, tonsil and adenoid removal surgery was performed, followed by surgery for otitis media at 5 years of age. During the most recent 3 hospitalizations, the child’s body weight, platelet count, hemoglobin levels, and coagulation function remained within normal ranges. On March 2020, at the age of 7 years, the child presented with a 1-day history of fever, accompanied by the presence of small bleeding spots on the skin and dark-colored urine. During a physical examination, the child exhibited a body mass index/age that was −0.56 standard deviations below the mean, indicating that her body mass index was lower than the average for her age. Additionally, her height/age was −2.13 standard deviations below the mean, suggesting that her height was significantly below the average for her age group. The examination also revealed mild jaundice, sparse hair, mild frontal bossing, low-set ears (Figure 1), and the presence of bleeding spots on the skin of the lower extremities.
During the hospitalization in March 2020, the child’s laboratory test results revealed several significant findings: hemoglobin level was 50 g/L; platelet count was 8×109/L; reticulocyte count was 29%; direct antiglobulin test was positive; plasma ADAMTS13 activity was 0%, with a positive plasma ADAMTS13 inhibitor; abnormal erythrocyte morphology in peripheral blood included the presence of erythrocyte fragments; activated partial thromboplastin time was prolonged; serum total bilirubin was elevated at 35 μmol/L (reference range: 5–21 μmol/L), with a high serum indirect bilirubin level of 30 μmol/L (range: 1–20 μmol/L); lactate dehydrogenase was markedly increased at 1528 U/L (range: 110–295 U/L); serum urea nitrogen was elevated at 14.75 mmol/L (range: 1.79–6.43 mmol/L); serum creatinine was within the reference range at 63 μmol/L (range: 5–77 μmol/L); antinuclear antibody (ANA) titer was 1: 40; and bone marrow cytology indicated the presence of a moderate number of megakaryocytes and poor platelet-producing function. Further, a psychological evaluation suggested mild intellectual disability.
A diagnosis of TTP and AIHA was given based on the clinical and laboratory data of the patient. During the hospitalization, the child received 10 mg/kg/day of methylprednisolone infusion for 5 days, followed by 2 mg/kg/day methylprednisolone, 2 g/kg immunoglobulin infusion, and transfusion of washed red blood cells and platelets. After the aforementioned procedures, there was an improvement in hemolysis; however, the platelet levels did not show significant improvement. Following 4 plasma exchanges, there was a successful increase in platelet production. However, there was also an observed tendency for platelet counts to decrease again. The treatment approach was then modified to rituximab at a dose of 375 mg/m2 per week. Notably, approximately 10 days after initiating this medication (Figure 2), the platelet counts gradually returned to normal levels.
After discharge, the patient maintained continuous oral administration of methylprednisolone (April 2020 to December 2022). During this period, the platelet count, hemoglobin levels, and coagulation function remained within normal ranges. However, after 3 hospitalizations for fever and the diagnosis of sepsis, there was a significant change in ANA levels, increasing from 1: 40 to 1: 1000. Laboratory tests during the last hospitalization in December 2022 indicated elevated ANA levels at 1: 1000, along with a high anti-double-stranded DNA level of 300 IU/mL (range: <100 IU/mL), and the presence of a positive anti-Smith antibody.
After the patient and family members signed the informed consent form for gene testing in March 2020, samples of 2 to 3 mL venous blood from all family members were collected and anticoagulated with EDTA. To determine the genetic cause in the proband, the present investigation encompassed karyo-type analysis, trio genome-wide copy number variation sequencing (CNV-seq), and trio whole-exome sequencing (trio-WES). DNA extraction was conducted using a fully automated nucleic acid extractor, and the quantification of DNA concentration was conducted using a NanoDrop 2000 nucleic acid concentration analyzer. The DNA concentration was found to be ≥50 ng/µL, and the A260/A280 purity ratio ranged from 1.8 to 2.0. Genome-wide CNV-seq was sequenced by means of Illumina Hiseq sequencers, and the total sequencing depth was approximately 0.2 to 0.6X (including areas with increased sequencing depth of approximately 1X-2X).Trio-WES was performed using the whole-exon oligonucleotide mircroarray, specifically the xGen Exome Research Panel v1.0, and it was sequenced using the Illumina NovaSeq 6000 sequencer. The sequencing coverage for the target sequence was ensured to be no less than 99%.
The library preparation process involved several steps, including gDNA ultrasonic fragmentation, end flattening repair, adapter ligation, magnetic-bead-based purification, PCR amplification, PCR product purification, and library quality assessment. The analysis of genome-wide CNV-seq involved an internal database analysis and screening. To assess the pathogenicity of CNVs, they were compared with pathogenic mutation databases such as Decipher and the general human genome database. In addition, a comprehensive analysis was conducted, considering dose sensitivity and the consistency of clinical features. The trio-WES analysis also involved an internal database analysis and screening. By combining pathogenic mutation databases, general human genome databases, clinical feature databases of 4000 known genetic diseases, and genetic data analysis algorithms, thousands of gene mutations were successfully graded. The mutation grade process was based on the 3-element grading system and the gene mutation grading system of the American College of Medical Genetics. Validation of the target sequence was conducted using Sanger sequencing on the ABI3730 sequencer following PCR. The results of the validation were acquired through the use of software that analyzes sequences.
The chromosome composition of the subject was 46,XX with the normal number of chromosomes. No clinically confirmed CNV mutations were found using the CNV-seq method (Figure 3). Trio-WES analysis revealed a mutation in the SHOC2 gene, specifically c.4 (exon2) A>G (p.Ser2Gly). This mutation was characterized by a heterozygous missense alteration in the second exon, leading to the replacement of serine with glycine at the second position within the SHOC2 gene (Figure 4). In addition, Sanger sequencing verified that both parents had a normal genotype at this site. Hence, this mutation represented a de novo mutation, and a similar pathogenic mechanism can be observed in autosomal-dominant diseases. Predictive assessments of functional protein damage resulting from the c.4 (exon2) A>G mutation in the SHOC2 gene of this patient, conducted using the SIFT, PolyPhen-2, and MutationTaster programs, consistently indicated that the mutation had a pathogenic effect. Based on the genetic mutation classification standards of the American College of Medical Genetics, the mutation site was determined to be a pathogenic variation (PS1+PS2+PM2+PP3). In summary, the identified missense mutation, non-frameshift mutation, or amino acid change aligned with the known pathogenic mutation (PS1). It was verified to be present in both parents (PS2), had a low-frequency variation with a minor allele frequency of less than 0.0005 (PM2), and was predicted to impact gene products by conservative protein-structure predictive software (PP3) (Table 2).
Discussion
The present findings suggest that the patient’s condition was closely related to NS/LAH but different from previous reports; MAHA and AIHA rarely co-occur in a patient [20–24]. From this case report we can learn that NS/LAH and RASopathy-associated developmental defects can lead to multisystem disorders in children.
NS/LAH is an autosomal-dominant RASopathy characterized by features that resemble NS in previously published reports [3–11]. The key distinguishing features in NS/LAH encompass ectodermal anomalies, such as loose anagen hair (thin, easily pluckable hair), a hypernasal voice, and hyperpigmented skin [12–15]. NS belongs to the group of conditions known as RASopathies, which result from gain-of-function mutations in genes involved in the RAS-MAPK signaling pathway [20]. NS typically presents in patients with distinct facial features, short stature, congenital cardiac defects, thoracic and skeletal anomalies, developmental delays, and bleeding disorders [21,22]. There have been 14 genes implicated in the etiology of NS, including PTPN11, KRAS, RIT1, SOS1, SOS2, RAF1, NRAS, BRAF, CBL, SHOC2, RRAS, MEK1, LZTR1, and PPP1CB [23,24].
SHOC2 gene mutations (c.4A>G, p.Ser2Gly) are common among patients with NS/LAH. The SHOC2 scaffold protein possesses an uncharacterized leucine-rich repeat domain. This protein is thought to play a role in localizing the protein phosphatase 1 catalytic subunit (PP1C) to the cell membrane [25].
The term RASopathy refers to a collection of multisystem disorders that clinically overlap with each other. Mutations in protein-encoding genes related to the RAS/MAPK cascade contribute to the development of one of the most notable sets of developmental disorders in humans [1,26]. To date, over 20 genes have been associated with RASopathies in scientific research [27]. The RAS pathway controls the progression of cell cycles, cellular expansion, reproduction, specialization, and metabolism [2,28,29]. Additionally, it has a vital function in the establishment and preservation of equilibrium in various tissue types. The RAS-MAPK pathway plays a crucial role in transmitting signals from outside the cell to promote cellular processes such as growth, development, viability, and metabolism. Following the binding of ligands, receptors on the cell surface undergo phosphorylation at specific locations within the cytoplasmic domain. Following activation, RAS proteins initiate the RAF-MEK-ERK cascade through a series of phosphorylation events, culminating in the translocation of activated ERK into the cell nucleus. The genetic dysregulation of ERK affects gene transcription and influences the function of the targets within the cytoplasm, resulting in cellular responses to the stimulus. Consequently, it is unsurprising that such dys-regulation can give rise to substantial clinical complications.
RASopathies can clinically manifest as a range of syndromes, including NS/LAH [13].
The patient in the present case was admitted to the hospital in March 2020 because of fever, thrombocytopenia, and anemia. At this time, a diagnosis of TTP combined with AIHA was given. The TTP in the patient was a typical MAHA [16], which was characterized pathologically by the formation of hyaline thrombi in the extensive microcirculation followed by ischemia in the associated tissue and organs [17]. The main clinical manifestations included the classic pentad of fever, thrombocytopenia, MAHA, renal impairment, and neurological deficits. Acquired TTP can be classified into 2 main categories: idiopathic TTP and secondary TTP. Idiopathic TTP constitutes the majority of cases and is linked to the production of ADAMTS13 antibodies within the body, for reasons that are currently unknown. On the other hand, secondary TTP is associated with abnormal endothelial damage triggered by factors such as infections, autoimmune disorders, pregnancy, medications, hematopoietic stem cell transplantation, and HIV, among others. The treatment for acquired TTP typically involves plasma exchange, often combined with immunomodulatory therapies, like corticosteroids, rituximab, splenectomy, and alacizumab. Rituximab is an effective treatment in this case. It is a chime-ric monoclonal antibody specifically targeting CD20 on mature B cells and exerts its therapeutic effect in TTP by depleting B cells, thereby suppressing the production of anti-ADAMTS13 autoantibodies and increasing ADAMTS13 activity [30,31].
The coexistence of MAHA and AIHA presents a diagnostic complexity due to the overlapping clinical features of these 2 conditions. One method to differentiate between the 2 conditions is by examining the morphology of red blood cells. AIHA is characterized by the presence of spherocytes, whereas schistocytes are typically found in MAHA. Additionally, AIHA results from the immune system’s attack on self-antigens present in erythrocytes, resulting in the untimely destruction of red blood cells [18]. The condition can be diagnosed through a positive direct antiglobulin test [19]. The positive direct antiglobulin test result in our patient, contrary to the typical findings in MAHA, indicated a mixed presentation of MAHA and AIHA.
After discharge, the child underwent continuous follow-up care for over 2 years, spanning from April 2020 to December 2022. With continued oral methylprednisolone treatment, the child maintained normal platelet counts and hemoglobin levels. However, there was a notable progressive increase in ANA levels, shifting from 1: 40 to 1: 1000, indicating a gradual development of immune dysregulation.
Autoimmune diseases are illnesses in which the immune system identifies and responds to self-antigens. There are 2 main categories of autoimmune diseases: organ-specific and systemic. Common autoimmune disorders comprise rheumatoid arthritis, type 1 diabetes mellitus, multiple sclerosis, Sjögren syndrome, and systemic lupus erythematosus [32]. In the preclinical phase of autoimmune disorders, there is an initial asymptomatic period of variable duration, followed by the emergence of nonspecific symptoms [33–35]. Before a clinical diagnosis is established, individuals with autoimmune conditions can experience a spectrum of autoimmune and inflammatory symptoms, which often worsen over the last months or years leading up to diagnosis.
A variety of autoimmune diseases have been reported among patients with RASopathy, including systemic lupus erythematosus, thyroiditis, and hepatitis [36]. In a clinical and serological study of 42 patients, there was a potential link identified between RASopathies and autoimmune diseases [37]. The findings from the study suggested that the acquired activation of the RAS/MAPK pathway in immune cells played a role in the progression of autoimmune disorders [36]. The complexity of the pathogenesis of autoimmune diseases has contributed to a shortage of effective treatments [32]. Therefore, it might be feasible to devise approaches for addressing autoimmune diseases by conducting a thorough investigation into the mechanisms that regulate SHOC2 and exploring strategies to target it in treatment.
Conclusions
SHOC2 mutations can potentially affect proteins involved in the RAS/MAPK pathway, leading to the development of RASopathies, which can clinically manifest as various syndromes, one of which is NS/LAH. Autoimmune diseases can develop due to RAS/MAPK pathway activation in immune cells. MAHA and AIHA rarely co-occur in a patient, especially in those with NS/LAH. The findings of the present study will help pediatricians expand their knowledge regarding NS/LAH and RASopathy-associated developmental defects that lead to multisystem disorders in children.
Figures
References:
1.. Zenker M, Clinical overview on RASopathies: Am J Med Genet C Semin Med Genet, 2022; 190(4); 414-24
2.. Roberts AE, Allanson JE, Tartaglia M, Gelb BD, Noonan syndrome: Lancet, 2013; 381(9863); 333-42
3.. Baldassarre G, Mussa A, Banaudi E, Phenotypic variability associated with the invariant SHOC2 c.4A>G (p.Ser2Gly) missense mutation: Am J Med Genet A, 2014; 164A(12); 3120-25
4.. Akgun-Dogan O, Simsek-Kiper PO, Taskiran E, ADA2 deficiency in a patient with Noonan syndrome-like disorder with loose anagen hair: The co-occurrence of two rare syndromes: Am J Med Genet A, 2019; 179(12); 2474-80
5.. Choi JH, Oh MY, Yum MS, Moyamoya syndrome in a patient with Noonan-like syndrome with loose anagen hair. Pediatr Neurol: Mar, 2015; 52(3); 352-55
6.. Cordeddu V, Di Schiavi E, Pennacchio LA, Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair: Nat Genet, 2009; 41(9); 1022-26
7.. Couser NL, Keelean-Fuller D, Davenport ML, Cleft palate and hypopituitarism in a patient with Noonan-like syndrome with loose anagen hair-1: Am J Med Genet A, 2018; 176(9); 2024-27
8.. Garavelli L, Cordeddu V, Errico S, Noonan syndrome-like disorder with loose anagen hair: A second case with neuroblastoma: Am J Med Genet A, 2015; 167A(8); 1902-7
9.. Gripp KW, Zand DJ, Demmer L, Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: Structural brain anomalies and myelofibrosis: Am J Med Genet A, 2013; 161A(10); 2420-30
10.. Kane J, Berrebi K, McLean R, Noonan syndrome with loose anagen hair associated with trichorrhexis nodosa and trichoptilosis: Clin Case Rep, 2017; 5(7); 1152-54
11.. Takasawa K, Takishima S, Morioka C, Improved growth velocity of a patient with Noonan-like syndrome with loose anagen hair (NS/LAH) without growth hormone deficiency by low-dose growth hormone therapy: Am J Med Genet A, 2015; 167A(10); 2425-29
12.. Motta M, Solman M, Bonnard AA, Expanding the molecular spectrum of pathogenic SHOC2 variants underlying Mazzanti syndrome: Hum Mol Genet, 2022; 31(16); 2766-78
13.. Patterson VL, Burdine RD, Swimming toward solutions: Using fish and frogs as models for understanding RASopathies: Birth Defects Res, 2020; 112(10); 749-65
14.. Wang Q, Cheng S, Fu Y, Yuan H, Case report: A de novo RASopathy-causing SHOC2 variant in a Chinese girl with noonan syndrome-like with loose anagen hair: Front Genet, 2022; 13; 1040124
15.. Mazzanti L, Tamburrino F, Scarano E, GH Therapy and first final height data in Noonan-like syndrome with loose anagen hair (Mazzanti syndrome): Am J Med Genet A, 2013; 161A(11); 2756-61
16.. Chao SH, Chang YL, Yen JC, Efficacy and safety of rituximab in auto-immune and microangiopathic hemolytic anemia: A systematic review and meta-analysis: Exp Hematol Oncol, 2020; 9; 6
17.. Joly BS, Coppo P, Veyradier A, Thrombotic thrombocytopenic purpura: Blood, 2017; 129(21); 2836-46
18.. Jager U, Barcellini W, Broome CM, Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev: May, 2020; 41; 100648
19.. Raos M, Lukic M, Pulanic D, The role of serological and molecular testing in the diagnostics and transfusion treatment of autoimmune haemolytic anaemia: Blood Transfus, 2022; 20(4); 319-28
20.. Bajia D, Bottani E, Derwich K, Effects of Noonan syndrome-germline mutations on mitochondria and energy metabolism: Cells, 2022; 11(19); 3099
21.. Nugent DJ, Romano AA, Sabharwal S, Cooper DL, Evaluation of bleeding disorders in patients with Noonan syndrome: A systematic review: J Blood Med, 2018; 9; 185-92
22.. Bruno L, Lenberg J, Le D, Novel approach to improve the identification of the bleeding phenotype in noonan syndrome and related RASopathies: J Pediatr, 2023; 257; 113323
23.. Athota JP, Bhat M, Nampoothiri S, Molecular and clinical studies in 107 Noonan syndrome affected individuals with PTPN11 mutations: BMC Med Genet, 2020; 21(1); 50
24.. Young LC, Hartig N, Boned Del Rio I, SHOC2-MRAS-PP1 complex positively regulates RAF activity and contributes to Noonan syndrome pathogenesis: Proc Natl Acad Sci USA, 2018; 115(45); E10576-E85
25.. Hauseman ZJ, Fodor M, Dhembi A, Structure of the MRAS-SHOC2-PP1C phosphatase complex: Nature, 2022; 609(7926); 416-23
26.. Kontaridis MI, Roberts AE, Schill L, The seventh international RASopathies symposium: Pathways to a cure-expanding knowledge, enhancing research, and therapeutic discovery: Am J Med Genet A, 2022; 188(6); 1915-27
27.. Tartaglia M, Aoki Y, Gelb BD, The molecular genetics of RASopathies: An update on novel disease genes and new disorders: Am J Med Genet C Semin Med Genet, 2022; 190(4); 425-39
28.. Tamburrino F, Scarano E, Schiavariello C, Endocrinological manifestations in RASopathies: Am J Med Genet C Semin Med Genet, 2022; 190(4); 471-77
29.. Gelb BD, Yohe ME, Wolf C, Andelfinger G, New prospectives on treatment opportunities in RASopathies: Am J Med Genet C Semin Med Genet, 2022; 190(4); 541-60
30.. Dane K, Chaturvedi S, Beyond plasma exchange: Novel therapies for thrombotic thrombocytopenic purpura: Hematology Am Soc Hematol Educ Program, 2018; 2018(1); 539-47
31.. Peyvandi F, Scully M, Kremer Hovinga JA, Caplacizumab for acquired thrombotic thrombocytopenic purpura: N Engl J Med, 2016; 374(6); 511-22
32.. Zhou F, Chen F, Ouyang Z, Functions of peroxiredoxins and their roles in autoimmune diseases: Antioxid Redox Signal, 2023 [Online ahead of print]
33.. Frazzei G, van Vollenhoven RF, de Jong BA, Preclinical autoimmune disease: A comparison of rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis and type 1 Diabetes: Front Immunol, 2022; 13; 899372
34.. Arbuckle MR, McClain MT, Rubertone MV, Development of autoanti-bodies before the clinical onset of systemic lupus erythematosus: N Engl J Med, 2003; 349(16); 1526-33
35.. Lu R, Munroe ME, Guthridge JM, Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies: J Autoimmun, 2016; 74; 182-93
36.. Uehara T, Hosogaya N, Matsuo N, Kosaki K, Systemic lupus erythematosus in a patient with Noonan syndrome-like disorder with loose anagen hair 1: More than a chance association: Am J Med Genet A, 2018; 176(7); 1662-66
37.. Quaio CR, Carvalho JF, da Silva CA, Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies: Am J Med Genet A, 2012; 158A(5); 1077-82
Figures
Tables
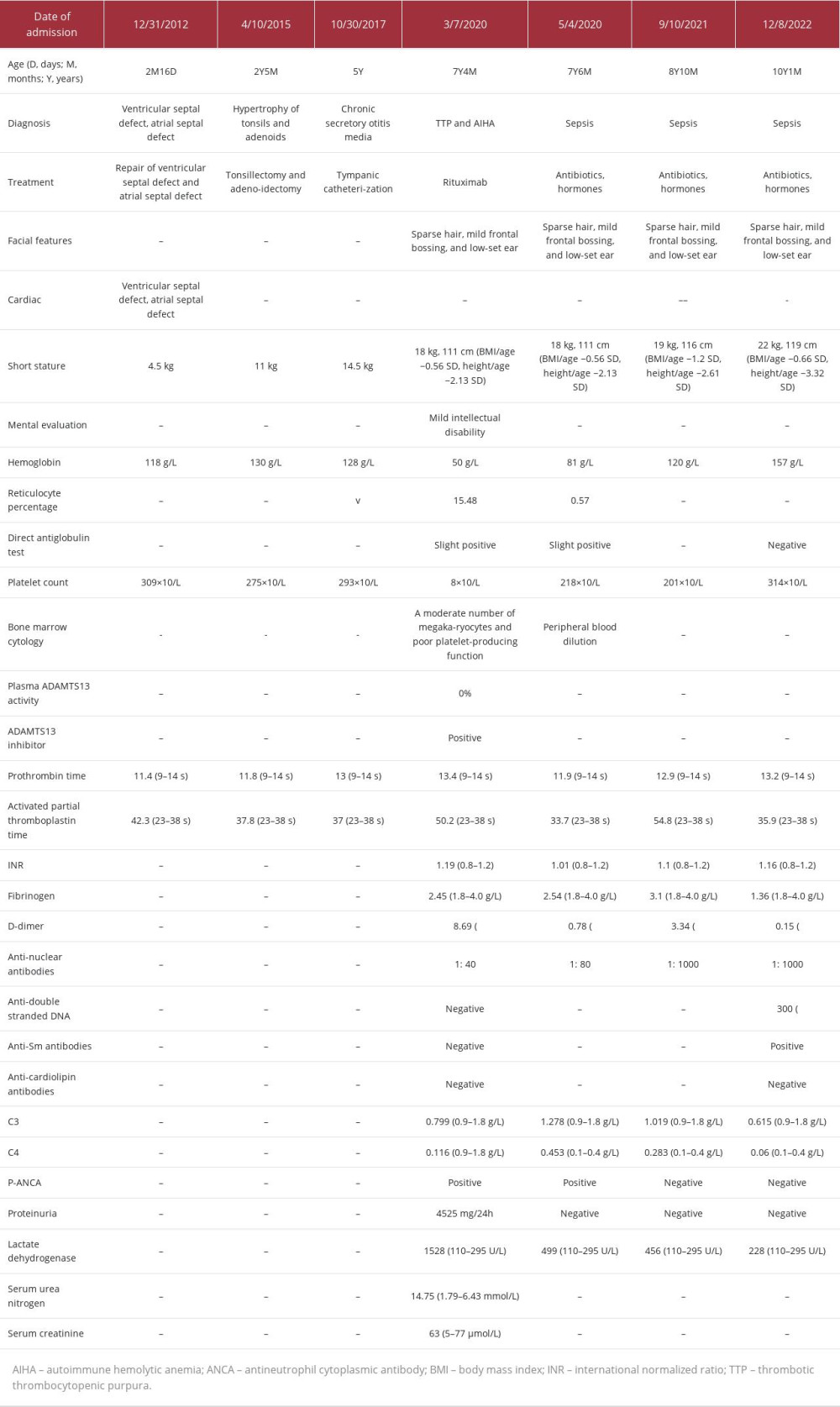 Table 1.. Summary of clinical information and 7 hospitalizations in our hospital over a 10-year period (December 2012 to December 2022).
Table 1.. Summary of clinical information and 7 hospitalizations in our hospital over a 10-year period (December 2012 to December 2022).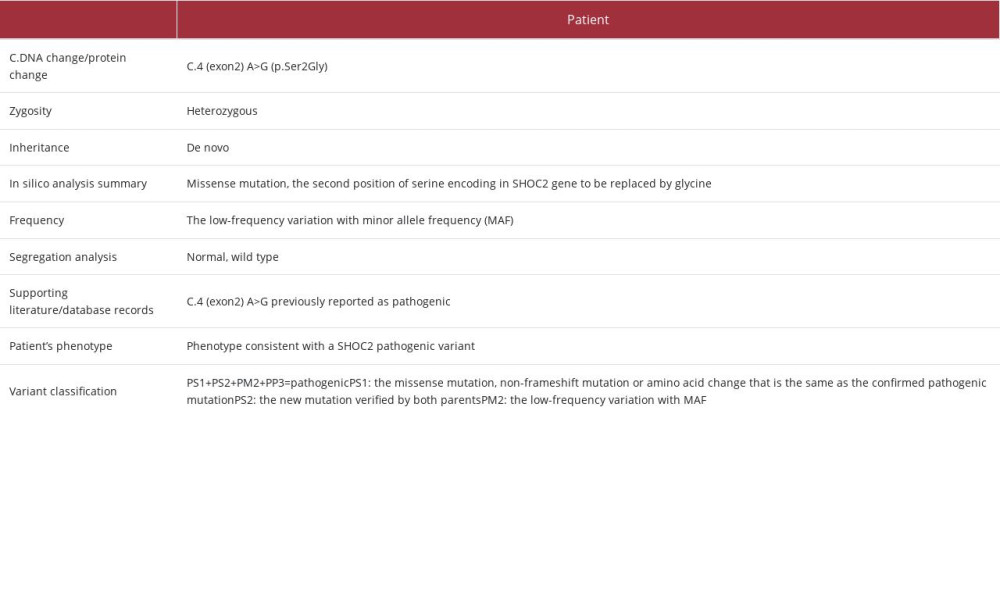 Table 2.. The SHOC2 variant interpretation for the patient.Table 1.. Summary of clinical information and 7 hospitalizations in our hospital over a 10-year period (December 2012 to December 2022).Table 2.. The SHOC2 variant interpretation for the patient.
Table 2.. The SHOC2 variant interpretation for the patient.Table 1.. Summary of clinical information and 7 hospitalizations in our hospital over a 10-year period (December 2012 to December 2022).Table 2.. The SHOC2 variant interpretation for the patient. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133