06 August 2025: Articles 
Unveiling Maternal Germline Mosaicism in X-Linked Alport Syndrome by Advanced Genetic Testing
Challenging differential diagnosis, Rare disease
Yuting Shen AEF 1,2, Caihong Liu BCD 2, Wei Wei BCD 2, Yongxiu HuangDOI: 10.12659/AJCR.947287
Am J Case Rep 2025; 26:e947287






Abstract
BACKGROUND: X-linked Alport syndrome is a hereditary disease caused by mutations in the COL4A5 gene, resulting in structural and functional abnormalities in the α5 chains encoded by these genes. This leads to the loss of type IV collagen in the basement membrane, causing dysfunction in organs such as the glomerulus, retina, and cochlea. In most cases, the COL4A5 pathogenic variant is inherited from a heterozygous mother in an X-linked dominant manner. However, in rare instances, XLAS can result from maternal germline mosaicism, where even though the mother’s somatic genetic testing detects no mutation, a subset of her germ cells carries a COL4A5 gene mutation, potentially leading to the birth of an affected child. This biological phenomenon can lead to the unexpected occurrence of affected offspring despite negative maternal carrier testing, with important implications for genetic counseling and recurrence risk assessment.
CASE REPORT: We report a case of maternal germline mosaicism in a family with X-linked Alport syndrome, where the proband – a 16-year-old male – carried a pathogenic variant in COL4A5 (NM_000495.5: exon 39: c. G3508A: p. G1170S). The proband’s sister also had the same variant and exhibited microscopic hematuria, while the mother showed no variant at this specific location, suggesting the presence of asymptomatic germline mosaicism.
CONCLUSIONS: Currently, few asymptomatic or mildly symptomatic females have been identified timely as carriers of germline mosaicism in X-linked Alport syndrome patients, warranting novel laboratory tools with high specificity, sensitivity, and minimal invasiveness for detecting mosaic phenomena, especially germline mosaicism.
Keywords: Nephritis, Hereditary, Chromosomal Instability, Mosaicism, Phenotype, Genotype, Humans, Collagen Type IV, Male, Female, Adolescent, genetic testing, Germ-Line Mutation, Pedigree
Introduction
Alport syndrome is an inherited disease caused by mutations in the
Case Report
The proband (II-1) was a 16-year-old male who presented with foamy urine for the past 4 months before admission. Urine analysis revealed 3+ proteinuria, with a 24-hour urinary protein concentration of 2.08 g, and 3–5 red blood cells per high-power field on microscopic examination. His serum creatinine level was elevated at 130.60 μmol/L, and renal ultrasound did not reveal any abnormalities. Blood pressure monitoring indicated hypertension at 150/90 mmHg. Renal pathology revealed irregular thickness, curvature, splitting, and lamination of the glomerular basement membrane, along with thickening of Bowman’s capsule and pericapsular fibrosis, which suggested a suspected diagnosis of hereditary kidney disease. Other tests such as auditory impedance, pure tone audiometry, and eye examination revealed no abnormalities.
Through next-generation sequencing (NGS) targeting glomerular disease-associated genes, a hemizygous missense variant of the
The proband’s father (I-1) died in an accident a few years ago and had no history of kidney abnormalities such as proteinuria or hematuria. To further investigate the mode of inheritance in the proband, urine analysis and blood tests were conducted for the proband’s mother (I-2) and sister (II-2) after informed consent. Interestingly, the proband’s sister showed minimal microscopic hematuria, with normal serum creatinine levels. Peripheral blood NGS targeting renal disease-associated genes revealed the presence of the same genetic variant (NM_000495.5: exon 39: c. G3508A: p. G1170S) in the proband’s sister (Figure 1B). However, the proband’s mother had normal urine analysis and renal function, and no variant was detected at this specific location (Figure 1C).
Discussion
The
The clinical phenotype of XLAS is influenced by the mode of inheritance and the sex of the affected individuals. The
The clinical presentation of XLAS is also influenced by the specific location and type of genetic variants [11].
Interestingly, as shown in Figure 2, a variant in the
Mosaicism refers to the presence of 2 or more different genotypes in an individual. Mosaicism arises when a mutation emerges in the early stages of embryonic development, resulting in a subset of cells possessing a distinct genetic makeup from the rest of the organism’s cells. Mosaicism can be categorized into somatic mosaicism, germline mosaicism, and mixed mosaicism, depending on the timing and distribution of genomic changes. Various mechanisms, such as chromosome nondisjunction, postzygotic lagging, and endoreduplication, can give rise to mosaicism. If the mutation occurs before the differentiation of primordial germ cells, it can be present in both germ cells and somatic cells. Due to different tissue involvement and varying degrees of mosaicism in each tissue, XLAS often exhibits different expression rates and incomplete penetrance [13]. Mosaicism is considered to be an important factor contributing to the milder phenotype observed in XLAS [14]. In the case mentioned, the proband’s mother did not display any signs of renal impairment, such as hematuria, proteinuria, or abnormal blood creatinine levels.
With advancements in genetic testing technologies, mosaic phenomena have been observed in various genetic diseases. Commonly used genetic testing methods include Sanger sequencing, targeted next-generation sequencing, and droplet digital PCR. These testing techniques analyze the proportion of mosaicism in somatic cells and can be performed using genomic DNA extracted from various sources, such as peripheral blood leukocytes, urinary sediments, hair roots, and skin. The presence of mosaic characteristics in the skin or kidneys of XLAS males may suggest the existence of mosaic phenomena. Identifying asymptomatic or mildly symptomatic females as carriers of germline mosaicism in XLAS patients is crucial for genetic screening in subsequent generations and assessing the risk of transmitting the disease during renal transplantation between parents [15]. However, detecting germline mosaicism is challenging because it requires testing germ cells. Current research is limited to studying germline mosaicism in males. Sampling for female germline mosaicism detection is invasive and ethically complex, making it difficult to determine the actual rate of germline mosaicism and the true recurrence risk.
Some scholars have attempted to infer the presence of germline mosaicism by screening for somatic mosaicism when suspecting its existence [16,17]. To date, only a few reports exist on sperm germline mosaicism in different genetic diseases. Using droplet digital PCR detection technology, Frisk et al discovered germline mosaicism in semen samples from patients without previously detected mosaicism in blood and observed that the level of mosaicism in sperm was always greater than that in blood [18]. However, there is no direct correlation between germline mosaicism and somatic mosaicism, and the detection of somatic mosaicism cannot directly predict the presence of germline mosaicism, nor can the abundance of somatic mosaicism be directly used to assess the abundance of germline mosaicism. In addition, researchers are working on the relationship between mosaicism modes and clinical phenotypes [19]. In XLAS patients, a correlation between the percentage of gene mosaicism and renal symptoms revealed a trend associating lower frequency of variant alleles with a lighter phenotype. In XLAS male patients with mosaicism, the renal immunohistochemistry showed a mosaic pattern, and the phenotype of renal symptoms was mild. XLAS women with germline mosaicism may not even have any associated clinical symptoms. Although their symptoms are mild, identification of gene mosaicism is crucial for family planning, as the risk for their offspring depends on the proportion of chimeras in the germline precursor cells. The susceptibility to mutations during germ cell development and the timing of mutations during parental embryogenesis may affect the abundance of mutations in both germline and somatic tissues, as well as the overall recurrence risk [20]. Analysis of tissues representing all 3 germ layers during embryonic development will aid in understanding when mutations occur and further deciphering the mechanism of mosaicism. Larger-scale studies of germline mosaicism in XLAS and other genetic diseases are needed to determine the exact recurrence risk.
Conclusions
We reported an identified variant (NM_000495.5: exon 39: c.G3508A: p.G1170S) in the
Figures
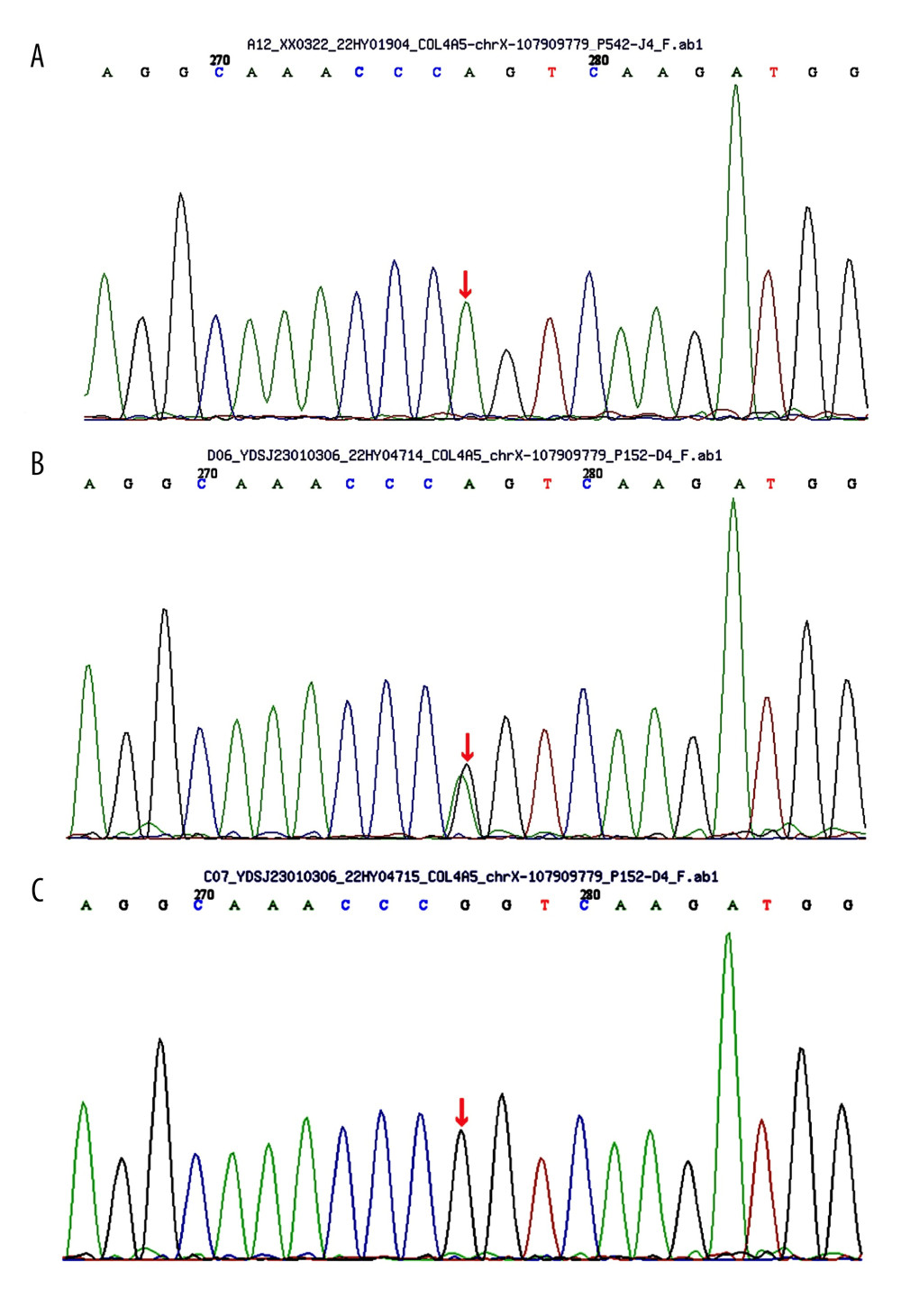 Figure 1. (A) Sequence of the heterozygous COL4A5 c.3508G>A p.G1170S del variant (II-1). (B) Sequence of the heterozygous COL4A5 c.3508G>A p.G1170S del variant (II-2). (C) Sequence of unaffected individuals (I-2).
Figure 1. (A) Sequence of the heterozygous COL4A5 c.3508G>A p.G1170S del variant (II-1). (B) Sequence of the heterozygous COL4A5 c.3508G>A p.G1170S del variant (II-2). (C) Sequence of unaffected individuals (I-2). 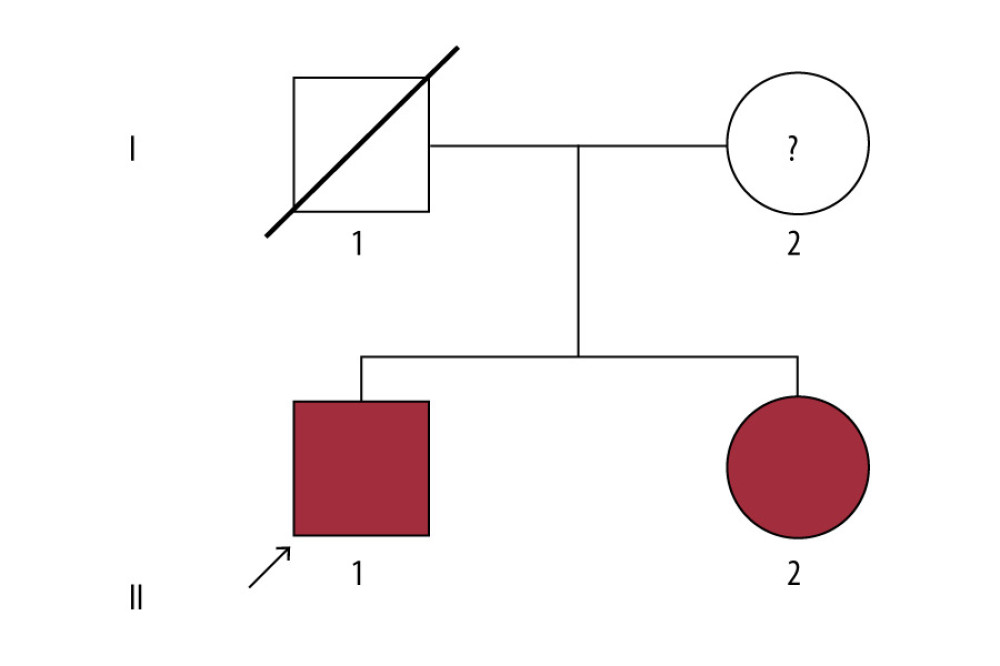 Figure 2. Pedigree of the Alport syndrome family. The red filled symbols represent patients carrying the COL4A5 c.3508G>A p.G1170S missense variant. The proband is marked with a black arrow.
Figure 2. Pedigree of the Alport syndrome family. The red filled symbols represent patients carrying the COL4A5 c.3508G>A p.G1170S missense variant. The proband is marked with a black arrow. References
1. Khoshnoodi J, Pedchenko V, Hudson BG, Mammalian collagen IV: Microsc Res Tech, 2008; 71(5); 357-70
2. Savige J, Storey H, Watson E, Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria: Eur J Hum Genet, 2021; 29(8); 1186-97
3. Nozu K, Nakanishi K, Abe Y, A review of clinical characteristics and genetic backgrounds in Alport syndrome: Clin Exp Nephrol, 2018; 23(2); 158-68
4. Gajecka M, Unrevealed mosaicism in the next-generation sequencing era: Mol Genet Genomics, 2015; 291(2); 513-30
5. Richards S, Aziz N, Bale S, Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology: Genet Med, 2015; 17(5); 405-24
6. Zhou J, Hertz JM, Leinonen A, Tryggvason K, Complete amino acid sequence of the human alpha 5 (IV) collagen chain and identification of a single-base mutation in exon 23 converting glycine 521 in the collagenous domain to cysteine in an Alport syndrome patient: J Biol Chem, 1992; 267(18); 12475-81
7. Kashtan CE, Ding J, Gregory M, Clinical practice recommendations for the treatment of Alport syndrome: A statement of the Alport Syndrome Research Collaborative: Pediatr Nephrol, 2013; 28(1); 5-11
8. Naito I, Kawai S, Nomura S, Relationship between COL4A5 gene mutation and distribution of type IV collagen in male X-linked Alport syndrome. Japanese Alport Network: Kidney Int, 1996; 50(1); 304-11
9. Savige J, Alport syndrome: Deducing the mode of inheritance from the presence of haematuria in family members: Pediatr Nephrol, 2020; 35(1); 59-66
10. Yamamura T, Nozu K, Fu XJ, Natural history and genotype–phenotype correlation in female X-linked alport syndrome: Kidney Int Rep, 2017; 2(5); 850-55
11. Singh SR, Savige J, Storey H, X-linked and autosomal recessive Alport syndrome: Pathogenic variant features and further genotype-phenotype correlations: Plos One, 2016; 11(9); e0161802
12. Savige J, Colville D, Rheault M, Alport syndrome in women and girls: Clin J Am Soc Nephrol, 2016; 11(9); 1713-20
13. Spinner NB, Conlin LK, Mosaicism and clinical genetics: Am J Med Genet C Semin Med Genet, 2014; 166(4); 397-405
14. Krol RP, Nozu K, Nakanishi K, Somatic mosaicism for a mutation of the COL4A5 gene is a cause of mild phenotype male Alport syndrome: Nephrol Dial Transplant, 2008; 23(8); 2525-30
15. Pinto AM, Daga S, Fallerini C, Detection of cryptic mosaicism in X-linked Alport syndrome prompts to re-evaluate living-donor kidney transplantation: Transplantation, 2020; 104(11); 2360-64
16. Deng H, Zhang Y, Ding J, Wang F, Detection of very low-level somatic mosaic COL4A5 splicing variant in asymptomatic female using droplet digital PCR: Front Med, 2022; 9; 847056
17. Okamoto T, Nozu K, Iijima K, Ariga T, Germline mosaicism is a pitfall in the diagnosis of “sporadic” X-linked Alport syndrome: J Nephrol, 2018; 32(1); 155-59
18. Frisk S, Wachtmeister A, Laurell T, Detection of germline mosaicism in fathers of children with intellectual disability syndromes caused by de novo variants: Mol Genet Genomic Med, 2022; 10(4); e1880
19. Fu XJ, Nozu K, Kaito H, Somatic mosaicism and variant frequency detected by next-generation sequencing in X-linked Alport syndrome: Eur J Hum Genet, 2016; 24(3); 387-91
20. Breuss MW, Yang X, Gleeson JG, Sperm mosaicism: Implications for genomic diversity and disease: Trends Genet, 2021; 37(10); 890-902
Figures
Figure 1. (A) Sequence of the heterozygous COL4A5 c.3508G>A p.G1170S del variant (II-1). (B) Sequence of the heterozygous COL4A5 c.3508G>A p.G1170S del variant (II-2). (C) Sequence of unaffected individuals (I-2).Figure 2. Pedigree of the Alport syndrome family. The red filled symbols represent patients carrying the COL4A5 c.3508G>A p.G1170S missense variant. The proband is marked with a black arrow. In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133