27 July 2025: Articles 
One Patient with 3 Antibody-Confirmed Neurological Autoimmune Syndromes: A Case Report and Review of the Literature
Challenging differential diagnosis, Management of emergency care, Clinical situation which can not be reproduced for ethical reasons, Rare coexistence of disease or pathology
Lauren A. Nguyen ABCDEF 1*, Frishan Rocel O. Paulo ABCDF 1, Jordan J. Petersen ABCDE 1, J. Douglas MilesDOI: 10.12659/AJCR.948329
Am J Case Rep 2025; 26:e948329






Abstract
BACKGROUND: The occurrence of multiple autoimmune neurological disorders in one patient is rare. Here, we present the case of a woman who exhibited clinical features and antibody titers consistent with myasthenia gravis (MG), neuromyelitis optica (NMO), and anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis.
CASE REPORT: A 37-year-old woman with a 10-year history of MG presented with a sudden loss of central vision in her right eye. Magnetic resonance imaging (MRI) revealed new enhancement of the right optic nerve, and additional cervical and thoracic spine scans showed continuous demyelination of the central cord. Given these findings, a primary demyelinating condition was suspected, and an NMO antibody test confirmed the diagnosis. Two years later, the patient developed significant behavioral changes, including neglecting her usual activities and displaying diminished responsiveness. She became mute and uncooperative with commands. Based on clinical suspicion of anti-NMDAR encephalitis, a comprehensive work-up revealed the presence of NMDAR antibodies, confirming the diagnosis. The patient was treated with plasma exchange, resulting in a marked improvement in her encephalopathy.
CONCLUSIONS: Over 12 years, this patient developed clinical manifestations of 3 distinct neurological autoimmune disorders. This case underscores the critical need for clinicians to remain alert to overlapping neurological conditions, enabling timely diagnoses and interventions that can help improve clinical outcomes and prevent unnecessary delays in treatment.
Keywords: Anti-N-Methyl-D-Aspartate Receptor Encephalitis, Autoimmune Diseases of the Nervous System, Myasthenia Gravis, Neurology, Neuromyelitis Optica, Humans, Female, adult, Autoantibodies, Magnetic Resonance Imaging
Introduction
Myasthenia gravis (MG), neuromyelitis optica (NMO), and anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis are rare autoimmune conditions characterized by neurological symptoms. In each, the underlying pathophysiology is the result of IgG autoantibodies. In the most common form of MG, IgG autoantibodies target the nicotinic acetylcholine receptor (AChR) at the neuromuscular junction, resulting in muscle weakness and rapid fatigue [1]. In NMO, autoantibodies bind to the membrane-bound water channel aquaporin-4 (AQP4), which is expressed on astrocytes. This leads to inflammatory lesions in the optic nerves and spinal cord, clinically manifesting as optic neuritis and transverse myelitis [2]. In anti-NMDAR encephalitis, autoantibodies target the GluN1 subunit of the NMDAR, leading to a reduction of receptor clusters at synapses, which manifests as psychiatric symptoms, cognitive disturbances, and movement disorders [3].
We present the case of a young woman diagnosed with MG, anti-NMDAR encephalitis, and NMO at different points in time. Each diagnosis was confirmed by specific antibody testing. It is known that individuals with MG have an increased risk of developing additional autoimmune disorders, compared with the general population, with the risk being highest among females and those with early-onset MG [4,5]. The coexistence of all 3 of these conditions has only been reported once previously, in a middle-aged woman who presented with a different sequence of diagnoses [6].
These cases merit consideration because they represent atypical manifestations of the individual conditions and demonstrate notable consistency in onset and progression. It has been suggested that the co-occurrence of these diseases may reflect an underlying predisposition to neurological autoimmunity or epitope spreading leading to the evolution of additional autoimmune responses [7,8]. Understanding these patterns and the related pathophysiology can inform treatment strategies, guide early screening, and support efforts toward prevention through early recognition and diagnosis.
Case Report
We report the case of a woman with a history of MG that had been previously diagnosed in her home country. She was 37 years old and had recently moved to the United States.
She first presented to our Emergency Department with fever, left-sided weakness, and an intermittent headache of 2 weeks duration that was accompanied by nausea and vomiting. A head computed tomography (CT) scan was performed, which revealed a left cerebellar hemorrhage with hemorrhagic components. During her hospitalization, she began to have a central vision loss in the right eye, with a “black circle” in the center of her vision. A new work-up was started, and an MRI showed right optic nerve enhancement, suggestive of an aggressive demyelinating process, when compared with a previous MRI ordered 1 day prior. MRIs of the cervical and thoracic spine showed continuous central cord demyelinating process, and suspicion for a primary demyelinating syndrome was high. The patient was started on methylprednisolone, and a titer for NMO antibodies, also known as AQP4 antibodies, came back positive after discharge (Table 1). The patient continued to demonstrate loss of central vision in the right eye but otherwise appeared stable. After the patient’s hospital discharge, she established outpatient care with a community neurologist.
We were unable to obtain the patient’s medical records from before she relocated to the United States. Her mother had Parkinson disease, but there was otherwise no known family history of neurological disease, and no known family history of autoimmune disease. At age 27 years, she presented to her family physician with a chief concern of blurred vision and vision loss. She was referred to a neurologist and MG was diagnosed, which was managed with pyridostigmine. Between ages 27 and 32 years, she had myasthenic crises 3 times. She underwent a thymectomy at age 32 years. At the time of her hospitalization at age 37 years, she was not on any steroid-sparing medication for her MG.
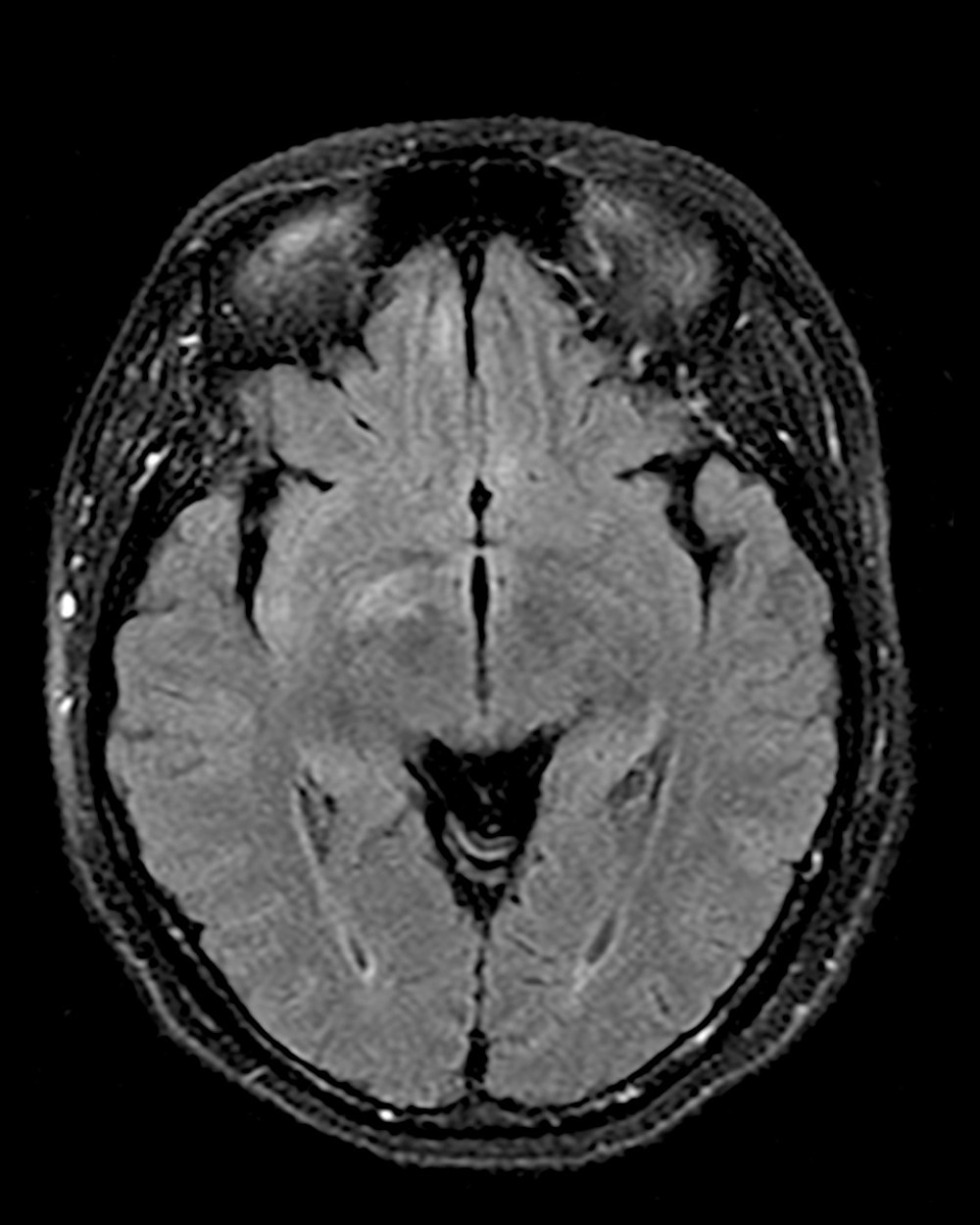
At age 39 years, 2 years after her initial hospitalization and diagnosis of neuromyelitis optica, the patient presented to the Emergency Department with altered mental status. She was accompanied by her husband, who revealed that she had been neglecting her usual household activities, including taking care of her children, and was experiencing periods of unresponsiveness, withdrawal, and urinary incontinence. On admittance to the hospital, she showed significant waxing and waning of her neurological status. Her Glasgow coma scale score varied from the minimum score of 3 to the maximum score of 15 over the course of an hour. On physical examination, she was mute and she was not following commands, although her pupils were reactive, visual fields were full, hearing was intact to conversation, and there was no facial weakness. She displayed occasional “seizure-like” behaviors consisting of lip smacking, her tongue protruding in and out of her mouth, spontaneous movements of her extremities, and her eyes rolling back. However, an electroencephalogram was negative for seizures. The rest of the physical examination was remarkable only for excess mucus secretions. An MRI of the brain was ordered, which revealed non-enhancing foci of T2 hyperintensity in the right caudate nucleus, bilateral medial inferior frontal lobes, and the right medial temporal lobe (Figure 1). Serum laboratory test results were negative for thyroid studies, electrolyte abnormalities, vitamin B9 (folate) or B12 (cobalamin) deficiencies, and MG (Table 2). The patient subsequently underwent 2 lumbar punctures. Cerebrospinal fluid (CSF) studies revealed a white blood cell count of 3 cells/μL, elevated IgG synthesis rate, absent oligoclonal bands, protein level of 41 mg/dL, a positive CSF-NMO antibody titer, serum NMO titer greater than 160 U/mL, and negative herpes simplex virus (Table 3). Notably, the CSF profile was also positive for NMDA antibodies (Table 3). A diagnosis of anti-NMDAR encephalitis was made. She was treated with plasma exchange. Her encephalopathy resolved, and a repeat MRI showed improvement of signal in the right caudate, insular cortex, and frontal lobes. While admitted, she developed pneumonia and was suspected to have a urinary tract infection. She was treated with tigecycline and meropenem. At the time of this admission, she was still not on immunosuppressive medication. Immunosuppressive medication, including rituximab, was discussed with the patient and her husband, but they wanted additional time to consider treatment options; therefore, she was discharged on pyridostigmine and referred to a new outpatient neurologist.
Since the diagnosis of this patient’s MG, NMO, and anti-NMDAR encephalitis, she has been treated with prednisolone. Despite this, she had 1 flare-up at age 45, when she presented with altered mental status. During this time, she was found to have severe visual impairment; pupils were not reactive to light on physical examination, and she did not see light or shadow. CT of the head without contrast demonstrated diffuse cerebral and cerebellar volume loss, which was worse than previous imaging. Her fever and altered mental status, likely secondary to a urinary tract infection, was treated and she was discharged.
After her discharge, the patient was seen by a physical therapist, who noted that she could no longer move or feel her lower extremities. This was a change, compared with her last hospitalization, when she was able to stand and walk with a walker 5 feet. The new neurological symptoms prompted a T-spine MRI, which showed new enhancing lesions in the T spine. This active demyelination was suggestive of a NMO exacerbation. Since the patient also had a gastric ulcer at the time, intravenous steroids were contraindicated, and the patient was started on plasma exchange. Plasma exchange was completed 10 days later, without significant improvement. The patient was then discharged, but continued to have severe weakness in the lower extremities and was still blind.
Discussion
Since the existence of an autoimmune disease implies a dysfunction in the immune system, it is not uncommon that more than one autoimmune disease can occur simultaneously [9]. It is estimated that 25% of patients with one autoimmune disorder can develop a second one during their lifetime [10]. The coincident occurrence of 3 or more autoimmune diseases in the same individual is known as multiple autoimmune syndrome (MAS) [9,11–13]. A specific mechanism underlying MAS has not been identified, but the involvement of both environmental and hereditary factors is suspected.
MAS is a rare condition, and the combination of MG, NMO, and NMDAR encephalitis is an unusual presentation of MAS. Each of these autoimmune diseases are rare individually, so their co-occurrence is significant. There have been documented case reports in which patients were diagnosed with 2 of these diseases, either simultaneously or sequentially. The most frequent combinations are the concurrence of NMO and NMDAR (NMO-NMDAR), and the combination of NMO and MG (NMO-MG) [7,8,14–17].
In cases of concurrent MG and NMO, patients are typically diagnosed with early-onset AChR antibody-positive MG several years before the onset of NMO, with AQP4 antibodies detectable in serum prior to clinical symptoms [14,15]. At the time of NMO diagnosis, MG is often clinically quiescent, and AChR antibody titers generally decline following MG treatment, including thymectomy [14].
Thymectomy, a common treatment for MG, has been implicated as a potential risk factor for the later development of NMO spectrum disorders (NMOSD). Ikeguchi et al [18] reported that 89% of patients with NMOSD had undergone thymectomy prior to disease onset, and Fattorossi et al [19] observed a reduction in regulatory T cells in MG patients who underwent thymectomy. Given the role of T cell–mediated immunity in NMOSD pathogenesis, these findings suggest that thymectomy can contribute to disease evolution through impaired immune regulation [20,21]. Recognizing this potential association is clinically important, as it underscores the need for vigilance in monitoring patients with MG after thymectomy, although the coexistence of MG and NMOSD does not appear to worsen prognosis [14].
Patients with NMO-NMDAR often exhibit atypical presentations or disease courses and require aggressive treatment [5–8]. Previous reports do not indicate a consistent pattern of one condition preceding the other [5–8]. Although the underlying mechanism remains unclear, accumulating evidence suggests a potential link between anti-NMDAR encephalitis and NMOSDs [8]. Despite their distinct clinical features, this overlap aligns with our patient’s case, in which psychiatric symptoms – including personality changes, unresponsiveness, and suspected depression – emerged 2 years after her NMO diagnosis. Given these findings, clinicians managing NMOSD should maintain a low threshold for testing anti-NMDAR antibodies in patients who later develop psychiatric symptoms.
To date, there are no documented cases of patients who are seropositive for NMDAR encephalitis and MG alone. In 2023, Schäfer, Christensen, and Jensen [22] described a case of a previously healthy 24 year-old man who developed first seronegative MG and then a few months later encephalitis with antibodies against AMPA and NMDA receptors. Further, there are many reports of MG concurrent with other forms of encephalitis, including anti-AMPA receptor encephalitis [23] and paraneoplastic encephalitis [24–26]. Notably, one case described a patient with MG and leucine-rich glioma-inactivated 1 (LGI1) protein antibody-associated encephalitis, a condition known for psychiatric symptoms similar to NMDA encephalitis, along with its hallmark faciobrachial seizures [27]. Our patient has not exhibited faciobrachial seizures and showed no signs of limbic involvement on imaging, making LGI1 encephalitis unlikely. Nevertheless, this case of concurrent MG and LGI1 encephalitis highlights the importance of considering this diagnosis when evaluating MG-positive patients presenting with encephalitic symptoms, such as altered mental status, personality changes, or seizures.
To date, only one documented case has described a patient with all 3 diagnoses confirmed by positive antibody assays [6]. Our patient shares similarities with this previously reported case, as both are female and initially presented to the Emergency Department with headaches and unilateral extremity weakness. However, they differ in the timing and sequence of their diagnoses. Our patient was diagnosed with MG at age 27 years, followed by NMO at 37 years, and NMDAR encephalitis at 39 years. In contrast, the patient described by Bonner et al [6] was first diagnosed with NMO at 44 years, then MG at 45 years, and finally NMDAR encephalitis 18 months later. Unlike our patient, who responded to first-line immunosuppressive therapy with glucocorticoids, the patient of Bonner et al [6] experienced subacute progressive cognitive decline, imbalance, and behavioral symptoms, including deficits in executive function, visuospatial skills, recall, and fluency on the Montreal Cognitive Assessment. After failing glucocorticoid therapy, she was treated with rituximab, which led to significant and sustained improvements in anxiety, behavioral symptoms, and cognition. Despite differences in disease progression and treatment response, both cases exhibited overlapping symptoms. In both patients, headaches and unilateral upper and lower extremity weakness contributed to an NMO diagnosis, while personality changes and altered mental status prompted the diagnosis of NMDAR encephalitis.
In contrast to many other documented cases of MAS, these cases are unusual in that all 3 of the autoimmune disorders affect the nervous system. This can present diagnostic challenges. For example, both MG and NMO can cause weakness. However, the patterns of weakness differ significantly. NMO would be expected to produce weakness in the extremities, particularly the lower extremities, without marked fluctuation and with hyperreflexia. MG can produce extremity weakness, but fluctuation is a hallmark of the disease, ocular weakness is very common, and hyperreflexia should prompt the clinician to look for another etiology. In the case described here, the onset of encephalopathy posed a diagnostic challenge and was considered to be possibly stemming from NMO. Maintaining a broad differential diagnosis and systematically addressing each possibility can help avoid a satisfaction of search error, as can identifying the previously unidentified comorbid condition.
While our patient responded to the first-line immunosuppressive therapy of glucocorticoids, the patient in the report by Bonner et al [6] failed to respond to glucocorticoids but responded well to rituximab. Rituximab, originally approved for the treatment of B-cell non-Hodgkin lymphoma in adults, is an anti-CD20 chimeric antibody with human IgG1 immunoglobulin that has been reported in previous literature to be increasingly adopted as an off-label treatment for all 3 overlapping autoimmune diseases in separate cases [28–30]. Once bound to CD20+ cells, rituximab is believed to induce cell death through mechanisms such as antibody-dependent cell-mediated cytotoxicity, antibody-dependent phagocytosis, and direct cellular effects. Additionally, it has been shown to disrupt B-cell maturation, further contributing to its immunomodulatory effects [31].
In NMOSDs, rituximab has been increasingly adopted as a first-line off-label treatment for patients, due to its ability to reduce the quantity of circulating B cells, which has been shown to have a pathogenic role in NMOSDs by producing anti-AQP4-IgG antibodies [32]. It has been suggested that early treatment with rituximab can reduce disability in NMOSDs [29]. The data suggesting rituximab’s clinical efficacy in treating MG is a bit murkier: it is currently regarded as a third-line option, and the benefit of rituximab therapy in MuSK-positive patients are more widely accepted than in AChR-positive seropositive patients [32,33]. For anti-NMDAR encephalitis, rituximab is typically regarded as a second-line immunotherapy, alongside cyclophosphamide. Studies have demonstrated that it can significantly improve outcomes in patients unresponsive to first-line treatment and help reduce relapse frequency [30]. Given its broad immunosuppressive effects across these conditions, rituximab presents itself as a promising therapeutic option for patients with concomitant autoimmune diseases, such as the case described here.
Conclusions
Over a span of 12 years, this patient sequentially developed clinical manifestations of 3 rare neurological autoimmune disorders. Not only did the symptoms of these conditions overlap, but the patient also tested seropositive for all 3 simultaneously. In this case, the patient responded well to plasma exchange and glucocorticoids, although other therapies, including rituximab, have been explored for treating each of these autoimmune diseases. This case highlights the clinical significance of recognizing that central (NMOSD and NMDAR encephalitis) and peripheral (MG) neurological syndromes can coexist or emerge at different times. Awareness of these overlapping conditions is crucial for prompt diagnosis and intervention, helping to prevent delays that could lead to worsened outcomes.
References
1. Fichtner ML, Jiang R, Bourke A, Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology: Front Immunol, 2020; 11; 776
2. Bukhari W, Barnett MH, Prain K, Broadley SA, Molecular pathogenesis of neuromyelitis optica: Int J Mol Sci, 2012; 13(10); 12970-93
3. Dalmau J, Armangué T, Planagumà J, An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models: Lancet Neurol, 2019; 18(11); 1045-57
4. Christensen PB, Jensen TS, Tsiropoulos I, Associated autoimmune diseases in myasthenia gravis. A population-based study: Acta Neurol Scand, 1995; 91(3); 192-95
5. Fang F, Sveinsson O, Thormar G, The autoimmune spectrum of myasthenia gravis: A Swedish population-based study: J Intern Med, 2015; 277(5); 594-604
6. Bonner K, Aboul Nour H, Memon AB, Overlapping autoimmune neurological syndrome: A case report of triple-positive antibody: Cureus, 2022; 14(9); e29379
7. Kruer MC, Koch TK, Bourdette DN, NMDA receptor encephalitis mimicking seronegative neuromyelitis optica: Neurology, 2010; 74(18); 1473-75
8. Ran Y, Wang L, Zhang F, Anti-NMDAR encephalitis followed by seropositive neuromyelitis optica spectrum disorder: A case report and literature review: Clin Neurol Neurosurg, 2017; 155; 75-82
9. Sarfaraz S, Anis S, Multiple autoimmune syndrome: An unusual combination of autoimmune disorders: Rev Recent Clin Trials, 2020; 15(3); 240-43
10. Mohan MP, Ramesh TC, Multiple autoimmune syndrome: Indian J Dermatol Venereol Leprol, 2003; 69(4); 298-99
11. Cojocaru M, Cojocaru IM, Silosi I, Multiple autoimmune syndrome: Maedica (Bucur), 2010; 5(2); 132-34
12. Kwong EYL, Kuok MCI, Chan WK, Case report: Multiple autoimmune syndrome (MAS) – an unusual combination: Front Pediatr, 2022; 10; 1054025
13. Humbert P, Dupond JL, The multiple autoimmune syndromes (MAS): Br J Dermatol, 1997; 136(3); 468-69
14. Leite MI, Coutinho E, Lana-Peixoto M, Myasthenia gravis and neuromyelitis optica spectrum disorder: A multicenter study of 16 patients: Neurology, 2012; 78(20); 1601-7
15. Kimura K, Okada Y, Fujii C, Clinical characteristics of autoimmune disorders in the central nervous system associated with myasthenia gravis: J Neurol, 2019; 266(11); 2743-51
16. Zhu Y, Wang B, Hao Y, Zhu R, Clinical features of myasthenia gravis with neurological and systemic autoimmune diseases: Front Immunol, 2023; 14; 1223322
17. Sinani AA, Maawali SA, Alshekaili J, Overlapping demyelinating syndrome (Neuromyelitis optica spectrum disorders NMOSD with anti-NMDA receptor encephalitis); A case report: Mult Scler Relat Disord, 2020; 42; 102153
18. Ikeguchi R, Shimizu Y, Suzuki S, Japanese cases of neuromyelitis optica spectrum disorder associated with myasthenia gravis and a review of the literature: Clin Neurol Neurosurg, 2014; 125; 217-21
19. Fattorossi A, Battaglia A, Buzzonetti A, Circulating and thymic CD4 CD25 T regulatory cells in myasthenia gravis: Effect of immunosuppressive treatment: Immunology, 2005; 116(1); 134-41
20. Cruz-Herranz A, Sagan SA, Sobel RA, T cells targeting neuromyelitis optica autoantigen aquaporin-4 cause paralysis and visual system injury: J Nat Sci, 2017; 3(5); e358
21. Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter: Ann Neurol, 2012; 72(1); 53-64
22. Schäfer J, Christensen PB, Jensen K, AMPA and NMDA receptor antibody autoimmune encephalitis preceded by ocular myasthenia gravis: A case report: BMC Neurol, 2023; 23(1); 102
23. Li X, Mao YT, Wu JJ, Anti-AMPA receptor encephalitis associated with thymomatous myasthenia gravis: J Neuroimmunol, 2015; 281; 35-37
24. George J, Salunkhe M, Bhatele P, Status epilepticus heralding thymomatous paraneoplastic multifocal cortical encephalitis in myasthenia gravis: Seizure, 2021; 93; 169-70
25. Miyazaki Y, Hirayama M, Watanabe H, Paraneoplastic encephalitis associated with myasthenia gravis and malignant thymoma: J Clin Neurosci, 2012; 19(2); 336-38
26. Hammoud K, Kandimala G, Warnack W, Vernino S, Multifocal paraneoplastic cortical encephalitis associated with myasthenia gravis and thymoma: Arch Neurol, 2009; 66(11); 1407-9
27. Wang M, Cao X, Liu Q, Clinical features of limbic encephalitis with LGI1 antibody: Neuropsychiatr Dis Treat, 2017; 13; 1589-96
28. Tandan R, Hehir MK, Waheed W, Howard DB, Rituximab treatment of myasthenia gravis: A systematic review: Muscle Nerve, 2017; 56(2); 185-96
29. Damato V, Evoli A, Iorio R, Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: A systematic review and meta-analysis: JAMA Neurol, 2016; 73(11); 1342-48
30. Titulaer MJ, McCracken L, Gabilondo I, Late-onset anti-NMDA receptor encephalitis: Neurology, 2013; 81(12); 1058-63
31. Davies A, Berge C, Boehnke A, Subcutaneous rituximab for the treatment of B-cell hematologic malignancies: A review of the scientific rationale and clinical development: Adv Ther, 2017; 34(10); 2210-31
32. Iorio R, Damato V, Alboini PE, Evoli A, Efficacy and safety of rituximab for myasthenia gravis: A systematic review and meta-analysis: J Neurol, 2015; 262(5); 1115-19
33. Vesperinas-Castro A, Cortés-Vicente E, Rituximab treatment in myasthenia gravis: Front Neurol, 2023; 14; 1275533
Tables
 Table 1. The patient’s initial serum laboratory values.
Table 1. The patient’s initial serum laboratory values. Table 2. The patient’s serum laboratory values from 2 years later, at her second admission.
Table 2. The patient’s serum laboratory values from 2 years later, at her second admission. Table 3. The patient’s cerebrospinal fluid laboratory test values from 2 years later, at her second admission.Table 1. The patient’s initial serum laboratory values.Table 2. The patient’s serum laboratory values from 2 years later, at her second admission.Table 3. The patient’s cerebrospinal fluid laboratory test values from 2 years later, at her second admission.
Table 3. The patient’s cerebrospinal fluid laboratory test values from 2 years later, at her second admission.Table 1. The patient’s initial serum laboratory values.Table 2. The patient’s serum laboratory values from 2 years later, at her second admission.Table 3. The patient’s cerebrospinal fluid laboratory test values from 2 years later, at her second admission. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133