17 December 2025: Articles 
COVID-19-Associated MDA5-Mediated Necrotizing Myositis
Challenging differential diagnosis, Unusual or unexpected effect of treatment, Rare disease
Sarah Steadman ABCDEF 1, Amit Sikder ABCDEF 2, Harsh R. DesaiDOI: 10.12659/AJCR.949788
Am J Case Rep 2025; 26:e949788






Abstract
BACKGROUND: Rhabdomyolysis and autoimmune myopathies often present with similar clinical features, including myalgias, muscle weakness, and elevated serum creatine kinase (CK) levels. This overlap can make early diagnosis challenging and delay appropriate management. The recent rise in SARS-CoV-2 infections has led to an increasing number of reports of autoimmune myositis associated with the virus.
CASE REPORT: Here, we present a unique case of a young African American male who developed myalgias and bilateral upper and lower extremity weakness without any skin changes following a recent SARS-CoV-2 infection. His CK level exceeded 300 000 U/L; however, he had normal inflammatory markers and a basic autoimmune panel, and a chest computed tomography scan did not demonstrate interstitial lung disease. Initial treatment with intravenous fluids for presumed viral-induced rhabdomyolysis showed minimal clinical improvement. Surprisingly, empiric steroid therapy led to a rapid recovery in both CK levels and symptoms. A subsequent muscle biopsy revealed necrotizing myositis, and an autoimmune myositis panel showed elevated melanoma differentiation-associated gene 5 (MDA5) antibodies.
CONCLUSIONS: To the best of our knowledge, this is the first reported case of MDA5-associated necrotizing myositis following SARS-CoV-2 infection. Most patients with MDA5-related disease develop skin changes and underlying interstitial lung disease; however, our patient lacked these findings. At an 8-month outpatient follow-up, the patient had residual proximal muscle weakness and remained on immunosuppressive therapy. Further research is needed to better understand the pathophysiology of COVID-19-mediated autoimmune myopathies and to develop targeted therapies for these patients.
Keywords: Autoimmune Diseases, COVID-19, myositis, rhabdomyolysis, Rheumatology, Humans, Male, Interferon-Induced Helicase, IFIH1, SARS-CoV-2, adult, Necrosis
Introduction
Rhabdomyolysis is a clinical syndrome characterized by muscle breakdown, typically presenting with myalgia, weakness, and dark urine due to myoglobinuria [1]. It is commonly triggered by trauma, drugs, or viral infections such as SARS-CoV-2, Epstein-Barr virus, HIV, and Coxsackie B virus [2,3]. Viral-induced rhabdomyolysis can result from direct myocyte injury or immune-mediated mechanisms [4–6]. Increasingly, viruses like SARS-CoV-2 have also been implicated in triggering autoimmune myopathies, including dermatomyositis and necrotizing myositis [7,8]. Distinguishing post-viral rhabdomyolysis from autoimmune myopathies can be challenging because of overlapping features, such as proximal muscle weakness and elevated creatine kinase (CK) levels [7,9]. While rhabdomyolysis typically improves with fluid resuscitation, autoimmune myopathies often require immunosuppressive therapy [10].
Idiopathic inflammatory myopathies are rare autoimmune conditions affecting skeletal muscle and include dermatomyositis, polymyositis, necrotizing myositis, and inclusion body myositis [11]. Necrotizing myositis presents with rapidly progressive, symmetrical weakness, elevated CK levels, and muscle biopsy findings of myofiber necrosis with minimal inflammation [12,13]. Antibodies typically involved in COVID-19-associated necrotizing myositis include the signal recognition particle, Ro52, and nuclear matrix protein 2 [14,15]. In contrast, antibodies against melanoma differentiation-associated protein 5 (MDA5) are typically linked to a distinct dermatomyositis subtype, characterized by skin lesions and a high risk of rapidly progressive interstitial lung disease [12,13]. MDA5 is a cytoplasmic RNA helicase involved in innate immunity; however, its role in necrotizing myositis has not been previously reported [16].
In this case report, we describe a rare presentation of MDA5 antibody–associated necrotizing myositis following SARS-CoV-2 infection. The initial clinical impression was post-viral rhabdomyolysis; however, failure to improve with supportive care prompted reconsideration of the diagnosis and empiric treatment with corticosteroids. The rapid clinical recovery with steroids prompted further immunologic evaluation, ultimately revealing isolated MDA5 antibody positivity and histologic confirmation of necrotizing myositis. To the best of our knowledge, this is the first reported case linking MDA5 antibodies to a necrotizing myopathy phenotype in the absence of classic dermatomyositis skin findings or interstitial lung disease. This case underscores the diagnostic complexity in distinguishing rhabdomyolysis from autoimmune myositis in the context of COVID-19 and highlights the importance of considering immune-mediated etiologies early in patients with an atypical disease or poor response to standard therapy.
Case Report
A 24-year-old African American male with no past medical history presented to the emergency department with progressive worsening of bilateral leg pain and mild shortness of breath. Four days prior to presentation, he developed a sore throat and fever and tested positive for SARS-CoV-2 using a home antigen test. Two days later, those initial symptoms resolved, but he then developed bilateral upper and lower extremities pain and weakness, along with dark-colored urine. He denied any recent strenuous exercise, travel, medication use, or any personal or family history of venous thromboembolism or autoimmune conditions. On review of systems, the only additional symptom he endorsed was unintentional weight loss over the past 2 months.
On initial review, he was found to have mild hypertension, with a blood pressure of 143/101 mmHg, mild tachycardia, with a pulse rate of 100 bpm, no fever, a respiratory rate of 18 breaths per minute, and oxygen saturation of 98% on room air. On physical examination, he had 4 out of 5 muscle strength in both the upper and lower extremities, with intact sensation and a painless range of motion. Cardiac, respiratory, abdominal, and integumentary examination findings were otherwise unremarkable. Further cardiac workup revealed a normal electrocardiogram and a negative initial troponin level. Computed tomography (CT) showed no evidence of acute cardiac, lung, or abdominal abnormalities. Laboratory test results were notable for markedly elevated muscle enzymes, including a CK level of 342 529 U/L (Table 1). Despite the profound CK elevation, he had no evidence of renal impairment; his initial serum creatinine level was 0.8 mg/dL and remained stable throughout his hospitalization (Table 1). Liver enzymes were elevated, but viral hepatitis testing was negative (Table 2); the transaminitis was presumed secondary to muscle injury. SARS-CoV-2 was confirmed by polymerase chain reaction testing. The urine drug screen and alcohol level were negative. Urinalysis revealed moderate proteinuria and a few granular casts and was positive for blood but contained very few red blood cells.
The patient was initially treated for presumed COVID-19-induced rhabdomyolysis with aggressive intravenous (IV) fluid rehydration using normal saline, targeting a urine output of 200 mL/h. Despite 3 days of IV hydration, there was no improvement in limb weakness or liver enzymes levels and only a minimal decline in CK levels. This lack of response raised concern for an underlying myositis – potentially viral, genetic, or autoimmune in origin. There was no evidence of glucose-6-phosphate dehydrogenase deficiency, and amino acid analysis showed mild deviations but did not indicate any known inborn error of metabolism per pathology review. Inflammatory markers, including C-reactive protein and erythrocyte sedimentation rate, were within normal limits. Preliminary autoimmune testing, including antinuclear antibody (ANA), antineutrophil cytoplasmic antibody (ANCA), scleroderma, and Sjögren panels, were all negative (Table 3). On hospital day 3, the patient was empirically started on a 3-day course of 1000 mg of IV methylprednisolone, which resulted in a marked improvement in proximal muscle strength. Given the unrevealing laboratory findings, a general surgery consult was obtained to assist with a muscle biopsy. A proximal right thigh muscle biopsy revealed necrotizing myopathy (Figure 1). By the day of discharge, the patient’s CK level had decreased to 2323 U/L, and his liver enzymes had trended down to aspartate transaminase 67 U/L and alanine aminotransferase 184 U/L. He was discharged on prednisone 1 mg/kg daily (maximum dose 80 mg), along with famotidine for stress ulcer prophylaxis, and scheduled for outpatient follow-up with rheumatology services.
At the outpatient rheumatology consultation, the patient underwent additional myositis-specific antibody testing (Table 3), which was notable for positive MDA5 antibodies. Repeat CT imaging did not reveal any evidence of interstitial lung disease. At the 8-month follow-up, the patient continued to report intermittent myalgias and demonstrated 4+ power in bilateral proximal upper and lower extremities. His CK level remained mildly elevated at approximately 300 U/L. He was tapered off steroids and continued treatment with mycophenolate mofetil and rituximab.
Discussion
In this report, we describe a complex and atypical case of necrotizing myositis in a young African American male patient following SARS-CoV-2 infection. COVID-19 has increasingly been recognized as a potential trigger for unmasking idiopathic inflammatory myopathies, likely through disruption of immune regulation, aberrant inflammatory responses, and exacerbation of muscle injury. However, cases of COVID-19-associated necrotizing myositis remain rare compared with other idiopathic inflammatory myopathy subtypes [14,15,17,18]. To the best of our knowledge, the role of anti-MDA5 antibodies in necrotizing myositis has not been previously reported, although their association with other inflammatory myopathies – particularly a distinct subtype of dermatomyositis – is well established. MDA5 is a pattern recognition receptor that can be activated by viruses such as SARS-CoV-2, leading to a robust innate immune response [8]. Specifically, MDA5 recognizes viral RNA and induces type 1 interferon production through activation of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway [19]. This pathway is normally regulated by a negative feedback loop [20]. In the presence of autoantibodies against MDA5, downstream signaling is dysregulated, resulting in overproduction of the type 1 interferons and clinical manifestations such as tissue inflammation and rapidly progressive interstitial lung disease [16].
An intriguing aspect of this case is the novel finding of MDA5-mediated necrotizing myositis. Notably, 80% to 100% of patients with MDA5-positive myositis develop symptoms early in the disease course. However, in this case, the patient did not exhibit any respiratory symptoms and lacked the typical pulmonary findings on chest CT imaging, which is uncommon in MDA5-positive disease. Studies have demonstrated a high mortality rate – ranging from 33% to 66% within 6 months – primarily due to respiratory failure from rapidly progressive interstitial lung disease, despite intensive treatment [12,13]. In contrast, our patient responded well to immunosuppressive therapy and experienced no respiratory complications. A study by Cao et al demonstrated a correlation between MDA5 antibody levels and disease severity [21]. It is possible that our patient’s case supports this theory, given his slightly positive MDA5 antibody result and relatively mild clinical course. However, the possibility of a false-positive MDA5 result cannot be excluded. Meta-analyses have shown that the sensitivity and specificity of MDA5 antibody testing vary depending on clinical presentation, with greater diagnostic accuracy in patients with rapidly progressive interstitial lung disease [22]. In contrast, our patient neither presented with interstitial lung disease symptoms nor developed them at the time of this report. Additionally, the relatively low antibody level could reflect a dampened serologic response due to the initiation of corticosteroid therapy prior to testing. This raises important questions about the timing of antibody testing and its interpretability in patients who have already received immunosuppressive treatment.
Our case illustrates the diagnostic challenge of distinguishing between rhabdomyolysis and autoimmune myopathies based on the initial clinical presentation. The patient showed minimal clinical improvement despite aggressive fluid therapy, prompting the care team to consider alternative etiologies. Surprisingly, initial laboratory investigations – including inflammatory markers and a basic autoimmune panel – did not yield any diagnostic clues. While his CK level began to downtrend by hospital day 3, possibly due to fluid therapy, his symptoms remained unchanged, leading the team to revise the treatment plan. Empiric initiation of corticosteroids resulted in rapid and marked clinical and biochemical improvement, raising suspicion for an underlying immunologic or inflammatory process, such as autoimmune myositis (Figure 2). A recent retrospective study by David et al examined cases of MDA5 activation following SARS-CoV-2 infection or vaccination [23]. Interestingly, the study found that most affected individuals were over 50 years of age and White – demographics not representative of our patient. Additionally, classic supportive findings, such as Raynaud phenomenon and positive ANA, were observed only in a subset of cases [23]. Notably, our patient’s renal function was preserved despite an exceptionally elevated CK level exceeding 300 000 U/L. This may be partly attributed to early, aggressive fluid resuscitation and is consistent with prior studies indicating that only about 46% of individuals with markedly elevated CK develop acute kidney injury [24]. The absence of overt clinical signs, symptoms, or definitive laboratory findings highlights the variability in disease presentation and underscores the difficulty of early recognition of autoimmune myositis, which can lead to delayed initiation of appropriate therapy.
This case raises important research questions: What genetic or immunological predispositions contribute to an increased likelihood of triggering an MDA5 autoimmune response? Is our patient an outlier, given his young age and absence of multisystem involvement, or does this reflect an under-recognition or under-testing of MDA5-associated necrotizing myositis in patients with similar clinical presentations?
Conclusions
This case of COVID-19-induced necrotizing myositis mediated by MDA5 highlights the atypical presentation of the condition. It underscores the need for heightened clinical vigilance when evaluating patients with nonspecific symptoms – such as muscle weakness and myalgias – in the context of viral infections. Given the growing recognition of viral-induced myositis, particularly in the setting of SARS-CoV-2, it is imperative that clinicians maintain a high index of suspicion for autoimmune etiologies in cases presenting as rhabdomyolysis. Early identification and intervention are critical for preventing severe complications, including acute kidney injury and long-term disability. This case also emphasizes the need for continued research into the pathophysiological mechanisms linking viral infections with myositis. A deeper understanding of these pathways may facilitate the development of targeted therapeutic strategies and improve patient outcomes.
Figures
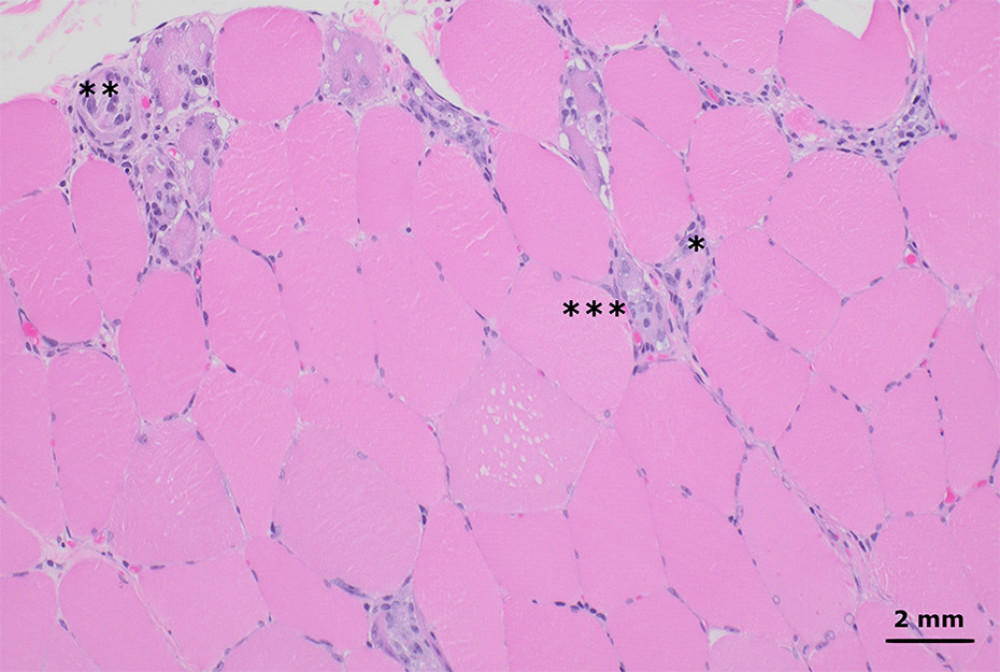 Figure 1. Hematoxylin and eosin-stained sections from formalin-fixed paraffin-embedded skeletal muscle tissue shows myofibers at different stages of degeneration and regeneration, including necrosis (*), myophagocytosis (**), and basophilic fibers (*** left of basophilic fibers). These changes affected a minority of the muscle tissue and were diffusely distributed throughout the muscle fascicles, without an obvious perifascicular pattern. There was no significant lymphocytic inflammation (200× total magnification).
Figure 1. Hematoxylin and eosin-stained sections from formalin-fixed paraffin-embedded skeletal muscle tissue shows myofibers at different stages of degeneration and regeneration, including necrosis (*), myophagocytosis (**), and basophilic fibers (*** left of basophilic fibers). These changes affected a minority of the muscle tissue and were diffusely distributed throughout the muscle fascicles, without an obvious perifascicular pattern. There was no significant lymphocytic inflammation (200× total magnification). 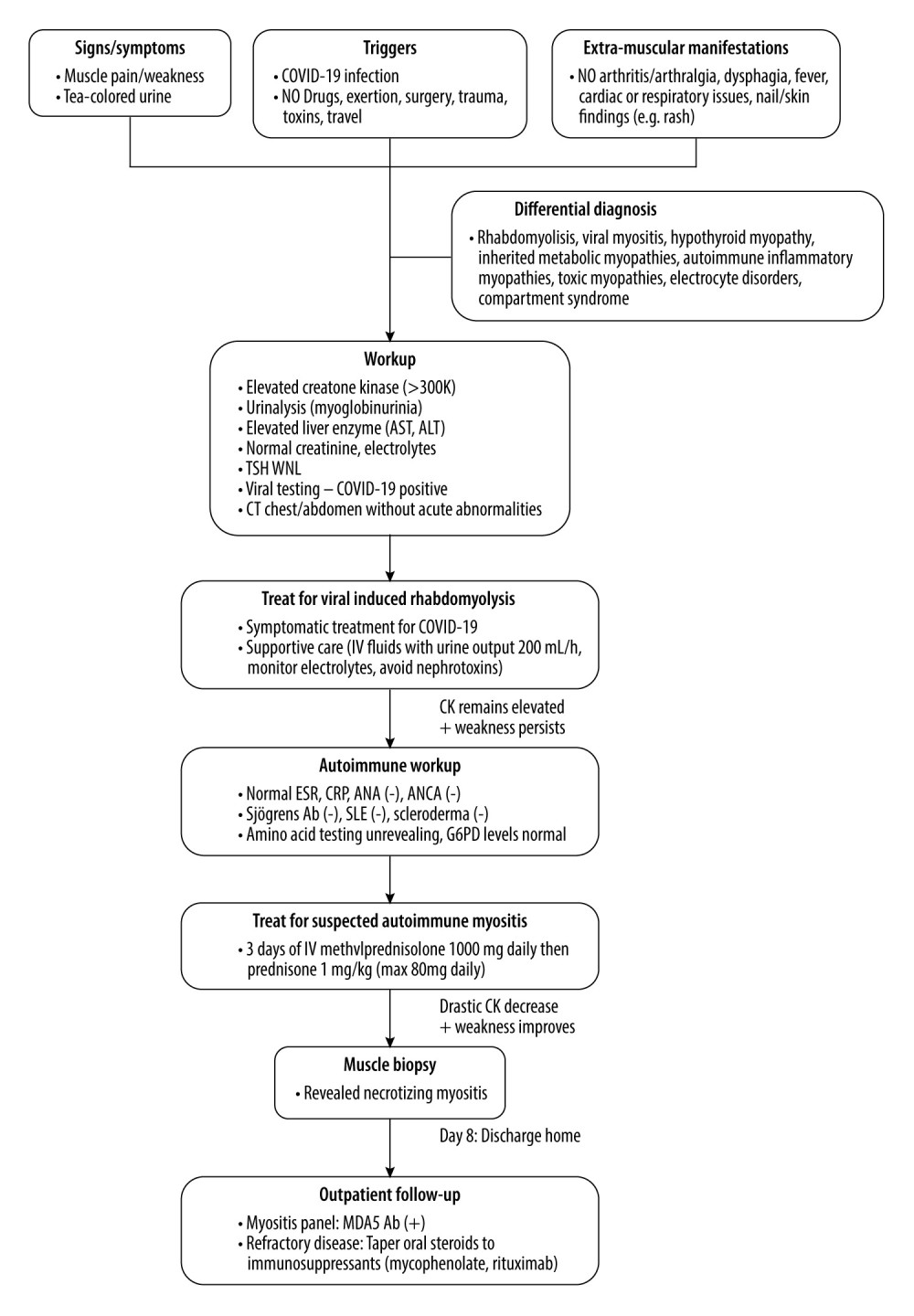 Figure 2. Flow diagram illustrating the decision-making process in diagnosing and managing this case.
Figure 2. Flow diagram illustrating the decision-making process in diagnosing and managing this case. References
1. Gupta A, Thorson P, Penmatsa KR, Gupta P, Rhabdomyolysis: Revisited: Ulster Med J, 2021; 90(2); 61-69
2. Long B, Koyfman A, Gottlieb M, An evidence-based narrative review of the emergency department evaluation and management of rhabdomyolysis: Am J Emerg Med, 2019; 37(3); 518-23
3. Preger A, Wei R, Berg B, Golomb BA, COVID-19-associated rhabdomyolysis: A scoping review: Int J Infect Dis, 2023; 136; 115-26
4. Patil P, Davidson J, Patel S, An undifferentiated cause of rhabdomyolysis: A case report: Int J Emerg Med, 2023; 16(1); 35
5. Dalakas MC, Polymyositis, dermatomyositis and inclusion-body myositis: N Engl J Med, 1991; 325(21); 1487-98
6. Johnson RW, Williams FM, Kazi S, Human immunodeficiency virus-associated polymyositis: A longitudinal study of outcome: Arthritis Rheum, 2003; 49(2); 172-78
7. Plavsic A, Arandjelovic S, Peric Popadic A, SARS-CoV-2-associated myopathy with positive anti-Mi-2 antibodies: A case report: Hong Kong Med J, 2023; 29(2); 170-72
8. Saud A, Naveen R, Aggarwal R, Gupta L, COVID-19 and myositis: What we know so far: Curr Rheumatol Rep, 2021; 23(8); 63
9. De Santis M, Isailovic N, Motta F, Environmental triggers for connective tissue disease: the case of COVID-19 associated with dermatomyositis-specific autoantibodies: Curr Opin Rheumatol, 2021; 33(6); 514-21
10. Sasaki H, Kohsaka H, Current diagnosis and treatment of polymyositis and dermatomyositis: Mod Rheumatol, 2018; 28(6); 913-21
11. Khoo T, Lilleker JB, Thong BYH, Epidemiology of the idiopathic inflammatory myopathies: Nat Rev Rheumatol, 2023; 19(11); 695-712
12. Wu W, Guo L, Fu Y, Interstitial lung disease in anti-MDA5 positive dermatomyositis: Clin Rev Allergy Immunol, 2021; 60(2); 293-304
13. Nombel A, Fabien N, Coutant F, Dermatomyositis with anti-MDA5 antibodies: Bioclinical features, pathogenesis and emerging therapies: Front Immunol, 2021; 12; 773352
14. Ibrahim A, Ghazali WSW, Misyail A, Immune-mediated necrotizing myopathy (Nam) related to SARS-Cov-2 infection: A case report: BMC Neurol, 2023; 23; 117
15. Loghman M, Rahmanian E, Alikhani M, Necrotizing autoimmune myositis following coronavirus disease 2019 infection: A case report: J Med Case Rep, 2022; 16(1); 488
16. Hu H, Yang H, Liu Y, Yan B, Pathogenesis of anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis: A concise review with an emphasis on type I Interferon System: Front Med, 2021; 8; 833114
17. Veyseh M, Koyoda S, Ayesha B, COVID-19 IgG-related autoimmune inflammatory necrotizing myositis: BMJ Case Rep, 2021; 14(4); e239457
18. Lokineni S, Mortezavi M, Delayed-onset necrotizing myositis following COVID-19 infection: Eur J Case Rep Intern Med, 2021; 8(4); 002461
19. Tonutti A, Motta F, Ceribelli A, Anti-MDA5 antibody linking COVID-19, type I interferon, and autoimmunity: A case report and systematic literature review: Front Immunol, 2022; 13; 937667
20. Yasukawa H, Sasaki A, Yoshimura A, Negative regulation of cytokine signaling pathways: Annu Rev Immunol, 2000; 18; 143-64
21. Cao H, Pan M, Kang Y, Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody: Arthritis Care Res, 2012; 64(10); 1602-10
22. Li L, Wang Q, Wen X, Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease: Oncotarget, 2017; 8(44); 76129-40
23. David P, Sinha S, Iqbal K, MDA5-autoimmunity and interstitial pneumonitis contemporaneous with the COVID-19 pandemic (MIP-C): EBioMedicine, 2024; 104; 105136
24. Bosch X, Poch E, Grau JM, Rhabdomyolysis and acute kidney injury: N Engl J Med, 2009; 361(1); 62-72
Figures
Figure 1. Hematoxylin and eosin-stained sections from formalin-fixed paraffin-embedded skeletal muscle tissue shows myofibers at different stages of degeneration and regeneration, including necrosis (*), myophagocytosis (**), and basophilic fibers (*** left of basophilic fibers). These changes affected a minority of the muscle tissue and were diffusely distributed throughout the muscle fascicles, without an obvious perifascicular pattern. There was no significant lymphocytic inflammation (200× total magnification).Figure 2. Flow diagram illustrating the decision-making process in diagnosing and managing this case. Tables
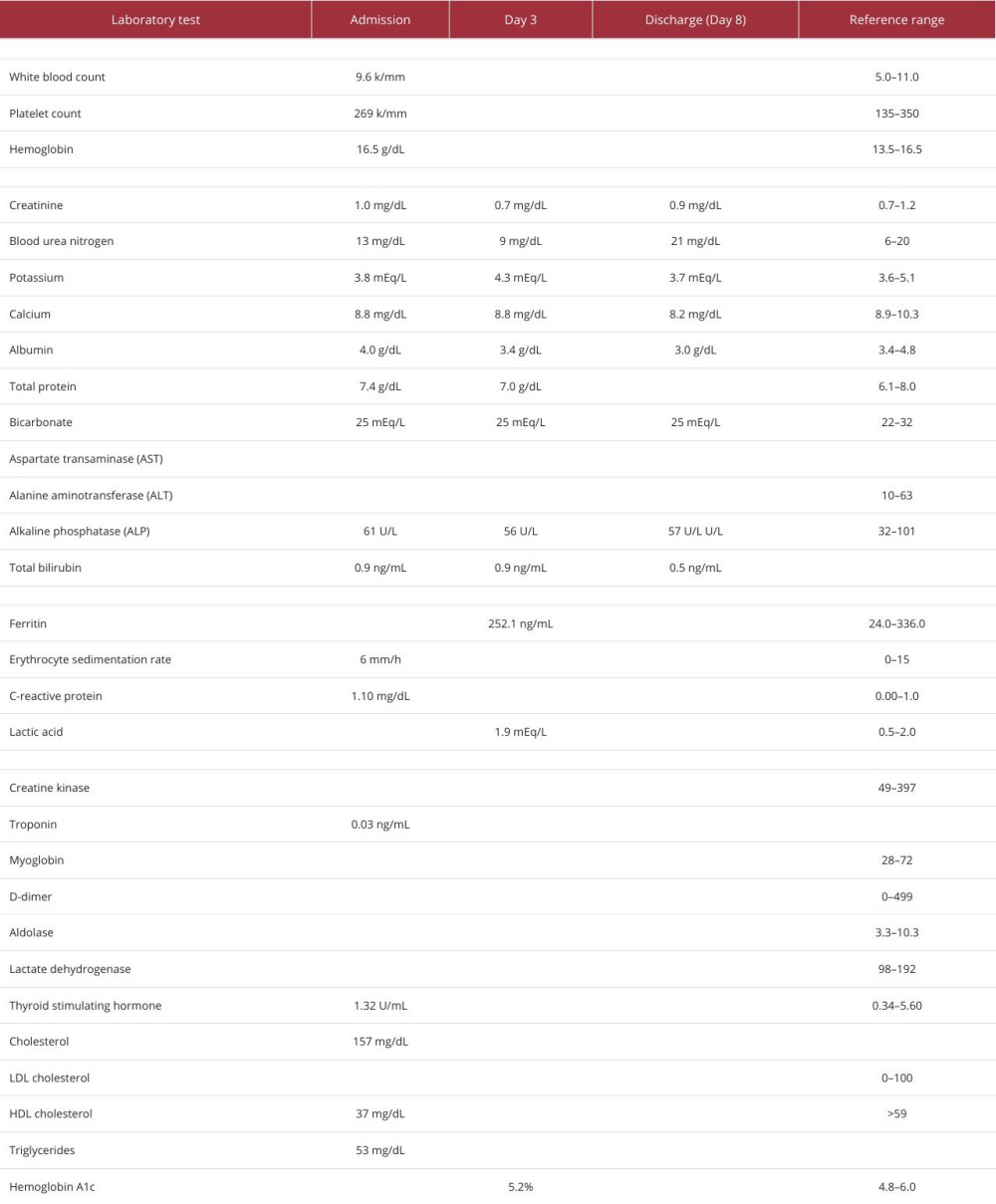 Table 1. Biochemical laboratory test values during hospital admission.
Table 1. Biochemical laboratory test values during hospital admission.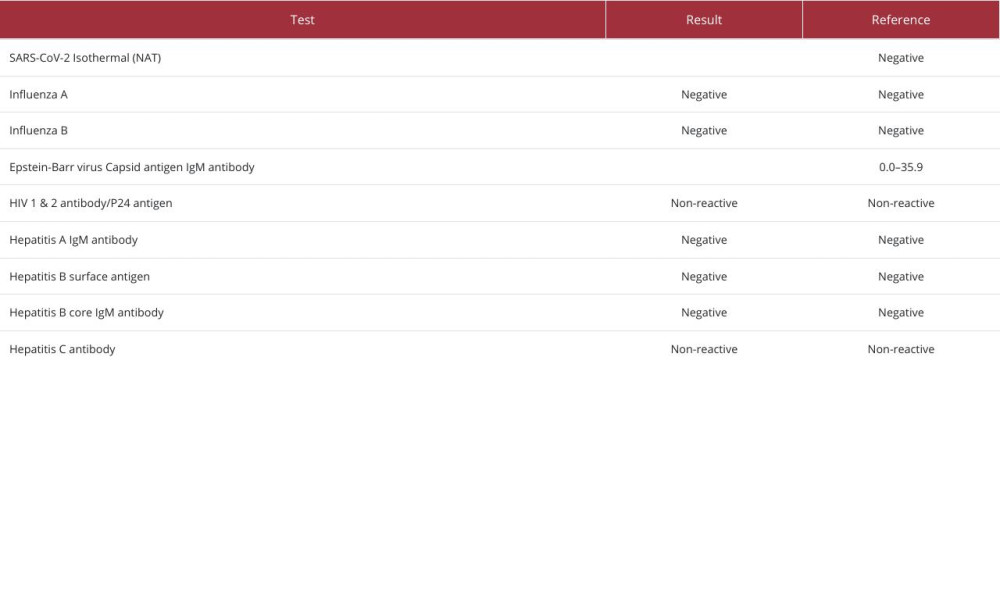 Table 2. Test results for viral infections during hospital admission.
Table 2. Test results for viral infections during hospital admission.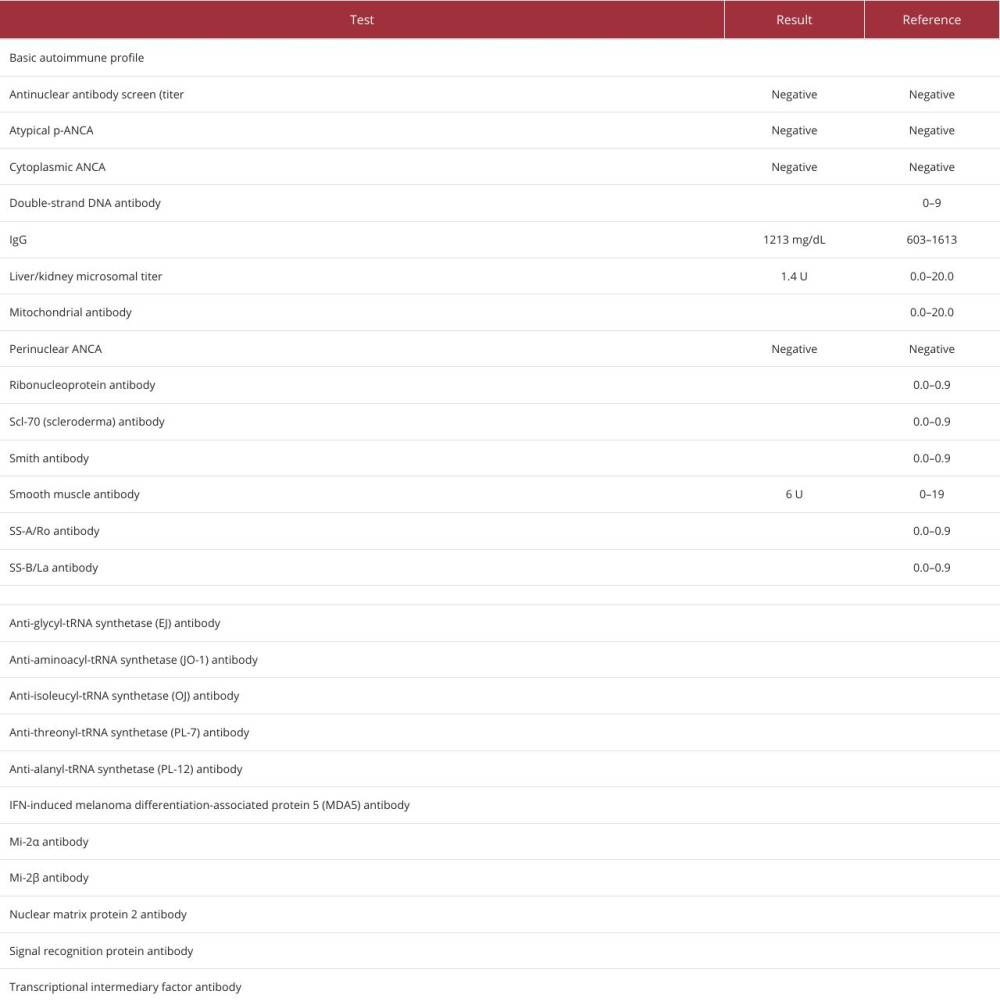 Table 3. Autoimmune testing performed during hospital admission and outpatient follow-up.Table 1. Biochemical laboratory test values during hospital admission.Table 2. Test results for viral infections during hospital admission.Table 3. Autoimmune testing performed during hospital admission and outpatient follow-up.
Table 3. Autoimmune testing performed during hospital admission and outpatient follow-up.Table 1. Biochemical laboratory test values during hospital admission.Table 2. Test results for viral infections during hospital admission.Table 3. Autoimmune testing performed during hospital admission and outpatient follow-up. In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133