14 December 2025: Articles 
Bruton’s Agammaglobulinemia: Case Series and Literature Review From King Fahad Central Hospital, Jizan, Saudi Arabia
Challenging differential diagnosis, Diagnostic / therapeutic accidents, Management of emergency care, Rare disease, Educational Purpose (only if useful for a systematic review or synthesis)
Ahmed Shamakhi ACDE 1, Nabil Dhayhi ACDE 2, Shatha Matabi BCF 3, Manal Maashi BCD 3, Abdullah Dhaifallah Hamdi BCDF 3, Bander Ali BC 4, Fadhel Hazazi DEF 5, Abdullah H. Alhamoud ABCDEF 3*DOI: 10.12659/AJCR.949936
Am J Case Rep 2025; 26:e949936






Abstract
BACKGROUND: X-linked agammaglobulinemia (XLA), also known as Bruton’s agammaglobulinemia, is a rare genetic disorder caused by mutations in the Bruton tyrosine kinase (BTK) gene. This defect leads to a substantial reduction in B cells and immunoglobulins, predisposing patients to recurrent severe infections. Early diagnosis and regular immunoglobulin replacement therapy are essential for effective disease management.
CASE REPORT: This report describes 2 male pediatric patients diagnosed with XLA at King Fahad Central Hospital in Jizan, Saudi Arabia. The first case involved a 14-month-old boy with recurrent respiratory infections, developmental delay, and frequent hospital admissions. Despite receiving immunoglobulin replacement therapy, his condition deteriorated due to poor treatment adherence, ultimately resulting in death. The second case involved a 3-year-old boy with recurrent respiratory infections and severe neutropenia. Genetic testing confirmed a BTK gene mutation. Consistent intravenous immunoglobulin therapy and close monitoring improved infection control and clinical stability, although he remained at risk for infections.
CONCLUSIONS: These cases emphasize the importance of early genetic diagnosis and strict adherence to immunoglobulin therapy in managing XLA. The differing outcomes underscore the critical role of treatment compliance in preventing severe complications and improving long-term prognosis. In regions with high consanguinity, genetic testing and family counseling are vital for early detection and optimal disease management.
Keywords: Agammaglobulinemia, Case Reports, Immunocompromised Host, infections, Humans, Male, Child, Preschool, Saudi Arabia, Genetic Diseases, X-Linked, Infant, Agammaglobulinaemia Tyrosine Kinase, Immunoglobulins, Intravenous, Fatal Outcome
Introduction
X-linked agammaglobulinemia (XLA), also known as Bruton’s agammaglobulinemia, is a primary immunodeficiency affecting the humoral immune response, first described by Bruton in 1952 [1]. The disorder results from mutations in the Bruton tyrosine kinase (
The prevalence of XLA varies by region, with approximately 250 cases per 100,000 men reported in Saudi Arabia and around 1 case per 190,000 men in the United States [6,7]. Diagnosis typically involves measuring serum immunoglobulin concentrations, enumerating B cells, and performing genetic testing to identify
This case series and literature review aim to describe the clinical and genetic characteristics of XLA observed at King Fahad Central Hospital in Jizan, Saudi Arabia, thereby contributing valuable data to the global understanding of this rare and serious immunodeficiency disorder.
Case Reports
CASE 1:
A 14-month-old male child had a history of recurrent chest infections and significant developmental delay; eventually, he was diagnosed with agammaglobulinemia via whole-exome sequencing. The analysis identified a hemizygous pathogenic
By 14 months of age, his condition had worsened. He was referred to a peripheral hospital after 6 days of fever, cough, and diarrhea. Despite treatment with antibiotics and antiviral agents, including oseltamivir, cefotaxime, and vancomycin, his clinical condition remained unstable. Eventually, he was hospitalized more than 5 times for respiratory infections, raising suspicion of agammaglobulinemia due to the infection frequency and severity.
Physical examination revealed edema of the lower limbs, hands, and periorbital regions, along with coarse facial features. His abdomen was distended; he exhibited cutaneous lesions and reduced air entry in the lower left lung. Laboratory investigations demonstrated severe immunodeficiency, with greatly decreased levels of all immunoglobulin classes: IgM levels ranged from 0.03 to 0.15 g/L (reference: 0.4–2.3 g/L), IgG levels from 0.03 to 13.55 g/L (reference: 7–16 g/L), and IgA levels from 0 to 0.08 g/L (reference: 0.7–4 g/L). Complete blood count results showed clinically significant abnormalities, including white blood cell counts fluctuating from 2.14 × 109/L to 18.64×109/L (reference: 4.5–10×109/L), persistently low hemoglobin levels around 6.3 g/dL (reference: 13–18 g/dL), and platelet counts varying from 12 to 563×109/L (reference: 150–400×109/L). He also had recurrent episodes of severe neutropenia, with neutrophil counts as low as 0.3×109/L (reference: 2.5–7.5×109/L), and elevated lymphocyte counts up to 13.69×109/L (reference: 1.5–3.5×109/L), further increasing his susceptibility to infections.
Imaging studies, including a computed tomography (CT) scan of the brain, revealed a subdural hygroma and mild cerebral atrophy (Figure 1). As the child grew older, adherence to the recommended monthly IVIG treatments declined, worsening his condition. At 2 years of age, he was admitted to the hospital with severe respiratory distress after missing 3 consecutive months of IVIG therapy due to social difficulties. He presented with fever, shortness of breath, and a worsening cough.
On examination, the patient was tachypneic (130 breaths/min), in respiratory distress, and exhibiting a distended abdomen with non-pitting edema of the lower limbs. Chest auscultation revealed bilateral wheezing and crackles. Despite aggressive management, including high-flow nasal cannula oxygen therapy, IVIG administration, and broad-spectrum antibiotics such as meropenem and vancomycin, his condition continued to deteriorate.
Throughout his illness, the patient experienced recurrent episodes of severe respiratory distress, frequently warranting intensive care. He was treated for multiple infections, including pneumonia, and required management of complications related to his immunodeficiency. Despite repeated interventions and the administration of IVIG, his prognosis remained poor. His clinical course was further complicated by inconsistent adherence to therapy, resulting in recurrent infections and repeated hospitalizations. Ultimately, the patient succumbed to his illness, primarily due to severe and recurrent respiratory infections associated with XLA and irregular treatment adherence.
The patient’s parents provided informed consent, verbally agreeing that the patient’s data could be used for this publication.
CASE 2:
A 3-year-old male child experienced repeated respiratory infections and severe neutropenia, leading to an eventual diagnosis of XLA. His history of repeated chest infections began at approximately 5 months of age, raising early suspicion of an underlying immunodeficiency. His first hospitalization was prompted by symptoms of gastroenteritis, including diarrhea and decreased oral intake, accompanied by severe neutropenia. Although he remained hydrated and alert, imaging studies (including CT scans) revealed bilateral pulmonary infiltrates with areas of consolidation (Figure 2) and ground-glass opacities consistent with pneumonia (Figure 3).
The diagnosis of agammaglobulinemia was confirmed through whole-exome sequencing, which identified a hemizygous
Throughout his clinical course, the patient experienced multiple severe infections, including nosocomial sepsis and urinary tract infections, requiring frequent hospitalizations and intensive treatment with broad-spectrum antibiotics such as vancomycin and tazocin. Despite regular IVIG therapy, difficulties in maintaining consistent adherence – particularly concerning timely IVIG administration – led to intervals of increased vulnerability to infection.
As the patient grew older, his condition stabilized to some extent with consistent IVIG therapy, although he continued to experience occasional respiratory infections. By the age of 4 years and 8 months, he was generally stable during follow-up visits, reporting no new complaints and exhibiting stable vital signs. Chest examinations remained clear, and he was active and afebrile. However, his clinical course was often disrupted by episodes of decreased appetite and activity, typically coinciding with acute infections. Despite these challenges, his management plan – which included regular IVIG therapy and close monitoring for infections – helped reduce the frequency and severity of his symptoms.
Although the patient’s outlook remained cautiously optimistic, the chronic nature of his condition and the recurrent infections requiring ongoing medical intervention complicated his clinical course. His experience underscored the importance of strict adherence to treatment regimens and continuous monitoring to prevent severe complications associated with XLA.
The patient’s parents provided informed consent, verbally agreeing that his data could be included in this publication.
Discussion
LIMITATIONS:
This case series is limited by its single-center design, which may restrict the generalizability of its findings. The small sample size of 2 cases also limits the ability to draw broader conclusions about XLA. Future studies with larger, multicenter cohorts and prospective designs are warranted to better assess long-term outcomes and optimize treatment strategies, particularly in regions with high consanguinity rates.
Conclusions
XLA presents substantial clinical challenges, primarily due to its association with recurrent severe infections. The cases described in this report highlight the critical roles of early diagnosis, consistent immunoglobulin replacement therapy, and close clinical monitoring in effective disease management. The differing outcomes observed underscore the importance of treatment adherence in preventing severe complications and improving long-term prognosis. Moreover, the identification of
Figures
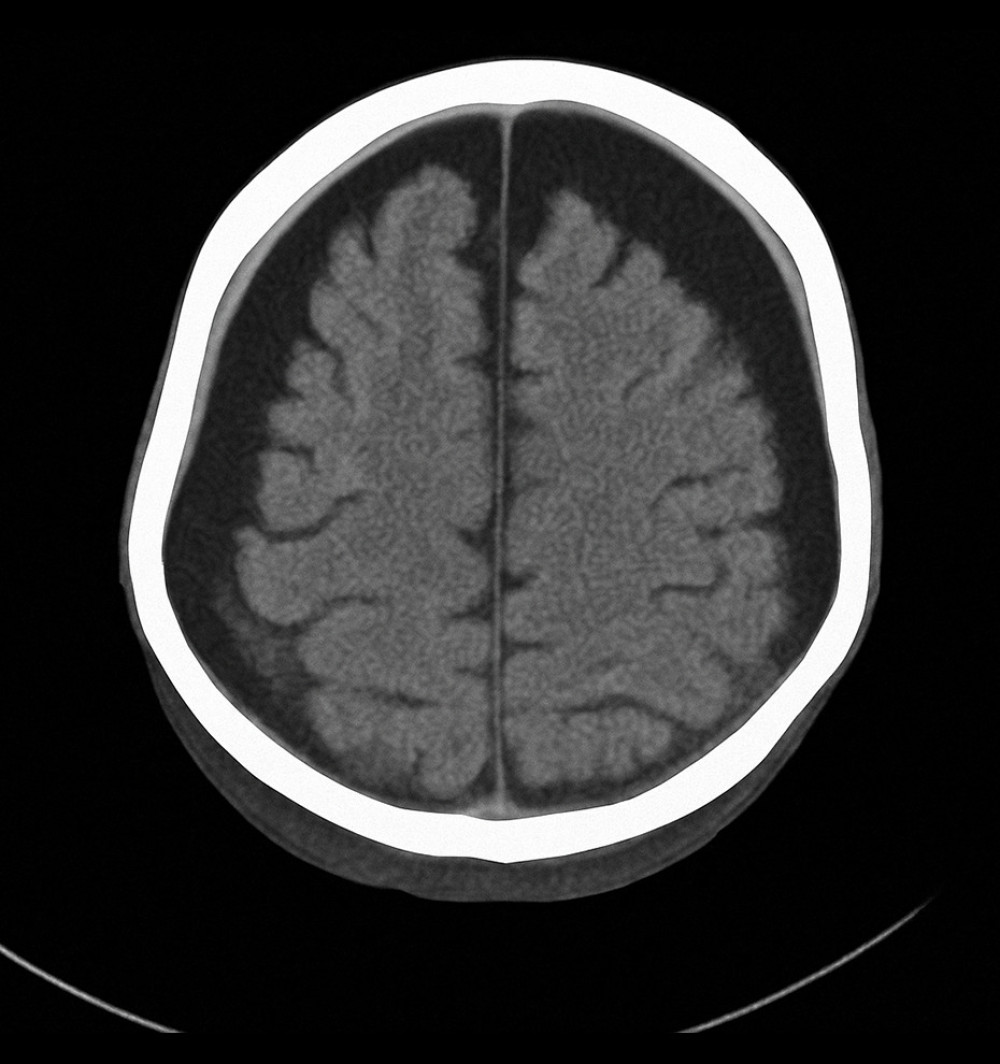 Figure 1. Axial computed tomography scan of the brain showing bilateral subdural hygroma and mild cerebral atrophy, consistent with chronic intracranial complications secondary to recurrent infections in X-linked agammaglobulinemia (Case 1).
Figure 1. Axial computed tomography scan of the brain showing bilateral subdural hygroma and mild cerebral atrophy, consistent with chronic intracranial complications secondary to recurrent infections in X-linked agammaglobulinemia (Case 1). 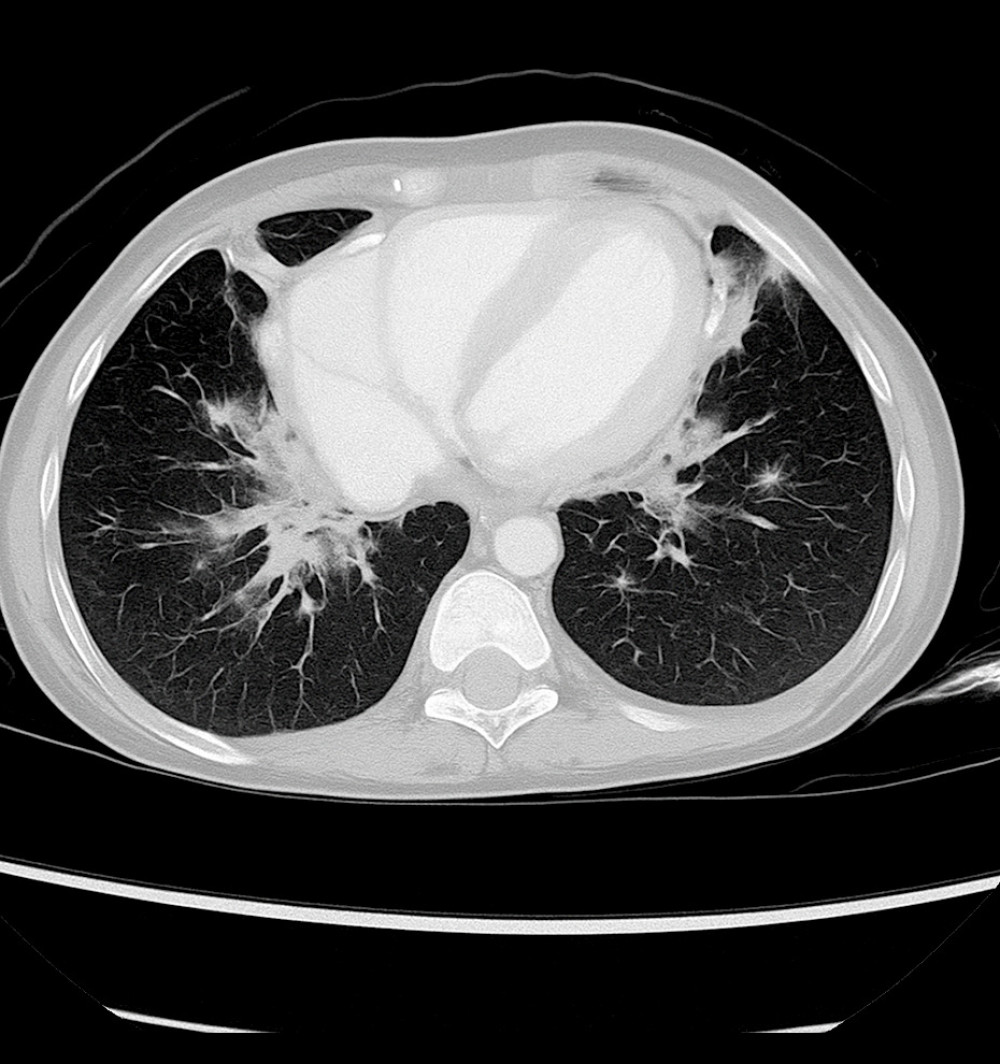 Figure 2. Axial chest computed tomography image showing bilateral pulmonary infiltrates with areas of patchy consolidation (Case 2).
Figure 2. Axial chest computed tomography image showing bilateral pulmonary infiltrates with areas of patchy consolidation (Case 2). 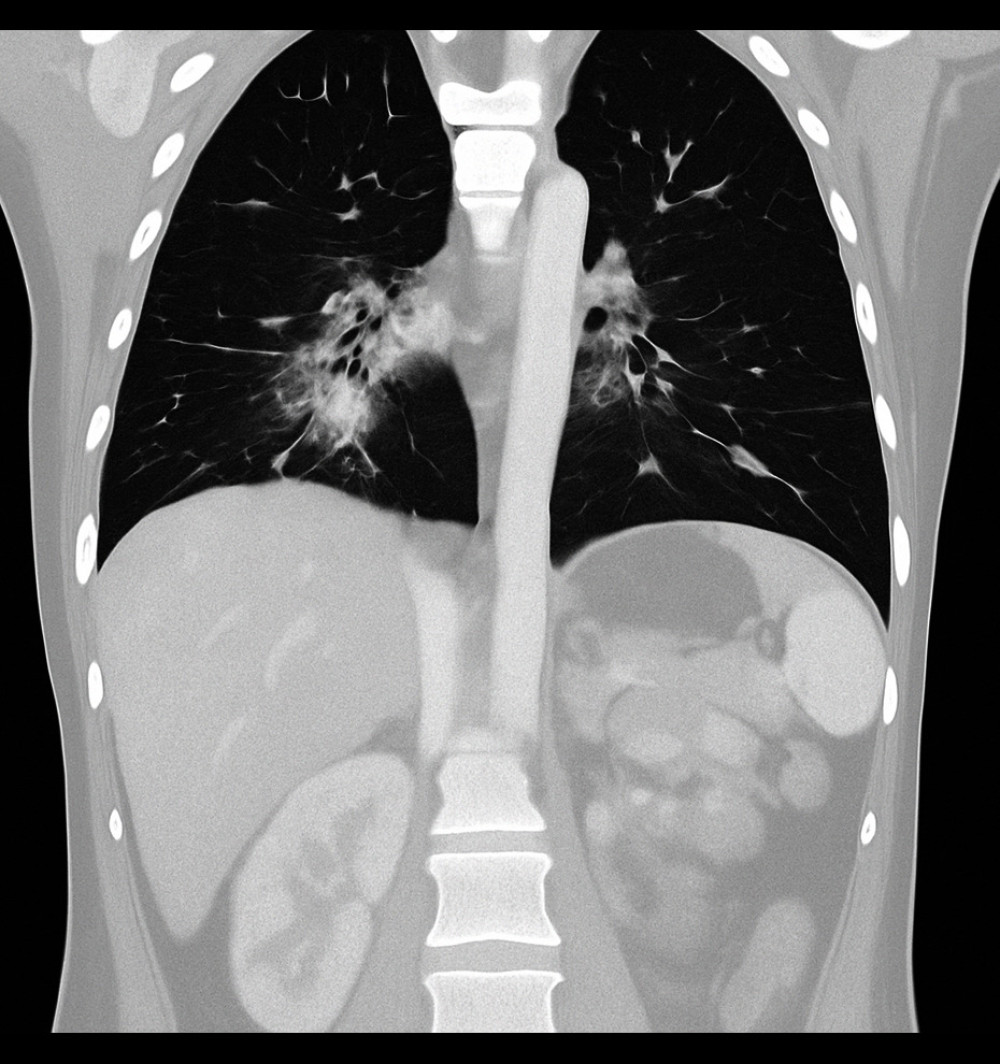 Figure 3. Coronal chest computed tomography image showing ground-glass opacities predominantly in the perihilar regions, consistent with an infectious or inflammatory process (Case 2).
Figure 3. Coronal chest computed tomography image showing ground-glass opacities predominantly in the perihilar regions, consistent with an infectious or inflammatory process (Case 2). References
1. Vetrie D, Vořechovský I, Sideras P, The gene involved in X-linked agammaglobulinemia is a member of the Src family of protein-tyrosine kinases: J Immunol, 2012; 188; 2948-55
2. Conley ME, Early defects in B-cell development: Curr Opin Allergy Clin Immunol, 2002; 2; 517-22
3. Ochs HD, Smith CI, X-linked agammaglobulinemia: A clinical and molecular analysis: Medicine (Baltimore), 1996; 75; 287-99
4. Lederman HM, Winkelstein JA, X-linked agammaglobulinemia: an analysis of 96 patients: Medicine (Baltimore), 1985; 64; 145-56
5. Shillitoe BMJ, Gennery AR, An update on X-linked agammaglobulinemia: Clinical manifestations and management: Curr Opin Allergy Clin Immunol, 2019; 19; 571-77
6. Al-Attas RA, Rahi AH, Primary antibody deficiency in Arabs: First report from eastern Saudi Arabia: J Clin Immunol, 1998; 18; 368-71
7. Taneja A, Muco E, Chhabra A, Bruton agammaglobulinemia: StatPearls [Internet], 2021, Treasure Island (FL), StatPearls Publishing [cited 2025 Oct 7]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK448170/
8. Holinski-Feder E, Weiss M, Brandau O, Mutation screening of the BTK gene in 56 families with X-linked agammaglobulinemia (XLA): 47 unique mutations without correlation to clinical course: Pediatrics, 1998; 101; 276-84
9. Conley ME, Broides A, Hernandez-Trujillo V, Genetic analysis of patients with defects in early B-cell development: Immunol Rev, 2005; 203; 216-34
10. Lindvall JM, Blomberg KE, Valiaho J, Bruton’s tyrosine kinase: Cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling: Immunol Rev, 2005; 203; 200-15
11. Väliaho J, Smith CI, Vihinen M, BTKbase: the mutation database for X-linked agammaglobulinemia: Hum Mutat, 2006; 27; 1209-17
12. Melo KM, Dantas E, De Moraes-Pinto MI, Primary immunodeficiency may be misdiagnosed as cow’s milk allergy: Seven cases referred to a tertiary pediatric hospital: ISRN Pediatr, 2013; 2013; 470286
13. Gajl-Peczalska KJ, Ballow M, Hansen JA, Good RA, IgE-bearing lymphocytes and atopy in a patient with X-linked infantile agammaglobulinemia: Lancet, 1973; 1; 1254
14. Ganier M, Lieberman P, Infantile agammaglobulinemia and immediate hypersensitivity to penicillin G: JAMA, 1977; 237; 1852-53
15. Al-Qadi B, Abu Taleb A, Salih S, Awareness of Jazan University students about consanguineous marriages and inherited disorders: Int J Health Sci, 2017; 5; 30-39
16. Conley ME, Rohrer J, Minegishi Y, X-linked agammaglobulinemia: Clin Rev Allergy Immunol, 2000; 19; 183-204
17. Smith CI, Islam TC, Bruton’s agammaglobulinemia: Clinical and genetic perspectives: J Immunol, 2012; 188; 1187-94
18. Viti R, Marcellusi A, Capone A, Direct and indirect costs of immunoglobulin replacement therapy in patients with common variable immunodeficiency (CVID) and X-linked agammaglobulinemia (XLA) in Italy: Clin Drug Investig, 2018; 38; 955-65
19. Lougaris V, Soresina A, Baronio M, Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality: J Allergy Clin Immunol, 2020; 146; 429-37
20. Kasahara Y, Imamura M, Shin C, Fatal progressive meningoencephalitis diagnosed in two members of a family with X-linked agammaglobulinemia: Front Pediatr, 2020; 8; 579
21. Melo KM, Alves LM, Valente CFC, Tavares FS, One-year intravenous immunoglobulin replacement therapy: Efficacy in reducing hospital admissions in pediatric patients with inborn errors of immunity: J Pediatr (Rio J), 2022; 98; 190-95
22. Lackey AE, Ahmad F, X-linked agammaglobulinemia: StatPearls [Internet[, 2025, Treasure Island (FL), StatPearls Publishing [cited 2025 Oct 7]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549865/
23. Xu Y, Qing Q, Liu X, Bruton’s agammaglobulinemia in an adult male due to a novel mutation: A case report: J Thorac Dis, 2016; 8; E1207-12
24. Zaidi SK, Qureshi S, Qamar FN, X-linked agammaglobulinemia: first case with Bruton tyrosine kinase mutation from Pakistan: J Pak Med Assoc, 2017; 67; 471-73
25. Al-Herz W, Chou J, Delmonte OM, Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population: Front Immunol, 2019; 9; 3146
26. Rawat A, Karuthedath Vellarikkal S, Verma A, Case report: Whole-exome sequencing identifies a novel frameshift insertion c.1325dupT (p.F442fsX2) in the tyrosine kinase domain of BTK gene in a young Indian individual with X-linked agammaglobulinemia: F1000Res, 2016; 5; 2667
27. Bedaiwy N, Alhamdi S, Al Suwairi W, Alsalamah M, Case report of a novel mutation in Bruton’s tyrosine kinase gene with confirmed agammaglobulinemia and absent B lymphocytes: LymphoSign Journal, 2022; 9; 1-4
Figures
Figure 1. Axial computed tomography scan of the brain showing bilateral subdural hygroma and mild cerebral atrophy, consistent with chronic intracranial complications secondary to recurrent infections in X-linked agammaglobulinemia (Case 1).Figure 2. Axial chest computed tomography image showing bilateral pulmonary infiltrates with areas of patchy consolidation (Case 2).Figure 3. Coronal chest computed tomography image showing ground-glass opacities predominantly in the perihilar regions, consistent with an infectious or inflammatory process (Case 2). In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952428
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952854
Most Viewed Current Articles
07 Dec 2021 : Case report  22,760,204
22,760,204
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,117
176,117
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,604
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,593
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133