23 August 2021: Articles 
Hepatic AL Amyloidosis without Significant Light Chain Elevation in a Patient Treated with CyBorD Plus Daratumumab
Unusual clinical course, Rare disease
Brad Rybinski1ABCDEFG*, Mehmet Kocoglu2ADEDOI: 10.12659/AJCR.933241
Am J Case Rep 2021; 22:e933241






Abstract
BACKGROUND: Immunoglobulin light chain (AL) amyloidosis is a plasma cell disorder in which excess light chain deposits in tissues, resulting in organ dysfunction and damage. Typically, AL amyloidosis presents as a systemic disease affecting multiple organs, and most patients have elevated serum free light chains. However, the presentation of AL amyloidosis is highly variable. The purpose of this case report is to raise awareness of the atypical presentations of AL amyloidosis in order to facilitate more rapid diagnosis, which has the potential to prevent additional organ damage when appropriate therapy is provided.
CASE REPORT: We describe a case of AL amyloidosis with amyloid deposition confined to the liver and bone marrow and lack of significant serum light chain elevation. The patient initially presented with a spontaneous hepatic hematoma, and was ultimately found to have hepatic AL amyloidosis, with monoclonal plasma cells in the bone marrow and monoclonal protein on serum protein electrophoresis. Our patient responded to treatment with cyclophosphamide-bortezomib-dexamethasone, the anti-CD38 antibody daratumumab, and autologous stem cell transplant, resulting in hematological and organ response.
CONCLUSIONS: AL amyloidosis can present with end-organ damage confined to isolated organs, and it can present without the expected elevation in serum light chains. Such patients can benefit from appropriate treatment, including traditional chemotherapy, daratumumab, and stem cell transplant. As effective treatments for AL amyloidosis are now available, prompt diagnosis has the potential to limit end-organ damage and potentially improve patient outcomes.
Keywords: Daratumumab, Immunoglobulin Light-chain Amyloidosis, Paraproteinemias, Amyloidosis, Antibodies, Monoclonal, Liver
Background
Immunoglobulin light chain (AL) amyloidosis is a hematological disorder in which a clonal population of plasma cells produces excess monoclonal light chain [1]. The excess light chain, which may be of lambda (λ) or kappa (κ) type, misforms into a β-pleated sheet and deposits into tissues, resulting in organ dysfunction [1]. Consequently, diagnostic criteria for AL amyloidosis require the presence of organ dysfunction secondary to amyloid deposition as identified by Congo Red staining, which detects the β-pleated sheet conformation, as well as demonstration by mass spectrometry or immunoelectro-microscopy that the amyloid is derived from light chains [2]. A monoclonal plasma cell proliferative disorder must also be present as evidenced by serum or urine monoclonal protein, abnormal free light-chain ratio, or clonal plasma cells in the bone marrow [2].
AL amyloidosis is a systemic disease that typically affects multiple organ systems, and typical presentations include renal impairment with nephrotic range proteinuria, diastolic heart failure with septal wall thickening, hepatomegaly with elevated alkaline phosphatase, and neuropathy [1,3]. Herein, we present an unusual case of AL amyloidosis in which end-organ damage was limited to the liver and bone marrow. The patient also lacked significant light chain elevation and responded to treatment with the anti-CD38 antibody daratumumab.
Case Report
A 60-year-old woman presented with spontaneous hepatic hematoma complicated by hemoperitoneum. The patient described waking up in the morning with acute onset, severe epi-gastric abdominal pain and non-bloody, non-bilious emesis. A few months prior, she had presented with a hepatic hematoma that failed initial conservative management and was treated by embolization. Neither hepatic hematoma was preceded by trauma. Additional past medical history was noncontribu-tory, and the patient had no history of cirrhosis, alcohol use disorder, or family history of bleeding or thrombosis. Imaging showed a large perihepatic hematoma of the lateral left hepatic lobe measuring 11.9×9 cm, with mass effect to the underlying hepatic parenchyma. The patient decompensated and developed severe abdominal distension with an acute drop in hemoglobin that did not respond to transfusion. The patient was taken to surgery for open left hepatectomy and exploratory laparotomy. Surgery encountered massive hemoperitoneum and extensive clot burden when entering the abdomen, and bleeding from the left hepatic vein was noted. Removal of hepatic segments 2 and 3 (corresponding to approximately half of the left lobe), the caudate lobe, and the gallbladder was required to control the bleeding, and bleeding from the hepatic vein itself was controlled surgically by oversewing. The liver was biopsied and stained positive with Congo Red (Figure 1A, 1B), consistent with a diagnosis of hepatic amyloidosis. The remainder of the hospital course was uncomplicated, and the patient’s hepatic function improved gradually during the hospitalization, rendering transplantation unnecessary and permitting safe discharge.
Workup for systemic amyloidosis was initiated. Liquid chromatography tandem mass spectrometry of the hepatic biopsy demonstrated a peptide profile consistent with AL (lambda)-type amyloid deposition. Intriguingly, the patient lacked significant light chain elevation at diagnosis. Serum free light chains showed kappa light chains of 12.11 (3.30–19.40 mg/L) and lambda light chains of 15.81 (5.70–26.30 mg/L), with a normal kappa/lambda ratio of 0.77 (0.26–1.65) (Table 1). Serum protein electrophoresis demonstrated a monoclonal protein of 0.63 g/dL, which was characterized as IgG lambda by immunofixation. Bone marrow biopsy showed a hypocellular marrow (5%) with tri-linage hypoplasia and diffuse staining for Congo Red within the interstitium and vascular walls, indicating amyloid deposition in the marrow. In addition, bone marrow biopsy was significant for lambda-restricted plasma cells (kappa: lambda ratio of 1: 4), with plasma cells comprising 5% of cellularity (MUM-1 stain). IgG was 1418 (700–1600 mg/dL), IgM was 25 (40–230 mg/dL), and IgA was 84 (70–400 mg/dL). All supporting diagnostic results are shown in Table 1. PET/CT whole-body imaging showed no evidence of plasmacytomas or lytic lesions.
We next focused on amyloidosis staging. A transthoracic echo-cardiogram demonstrated normal septal wall thickness and ventricular function (left ventricular ejection fraction 70%), NT-proBNP was 805 pg/mL (normal for age <900 pg/ml), and Troponin I was not elevated, ruling out cardiac amyloidosis. Endoscopic biopsy and Congo Red staining showed no evidence of amyloid deposition in the GI tract. There was no decrease in GFR or evidence of nephrotic syndrome. A physical exam showed no neurological findings, macroglossia, or pseudohypertrophy. Consequently, there was no evidence of amyloidosis affecting organs other than the liver and bone marrow in our patient.
On initial presentation, before the patient decompensated, hepatic biomarkers were mildly elevated: AST was 75 (14–36 U/L), ALT was 42 (0–34 U/L), alkaline phosphatase was 309 (38–126 U/L), and total bilirubin was 0.9 (0.3–1.2 mg/dL). Transaminases and total bilirubin rose markedly during our patient’s admission, reflecting the hepatic hematoma, but gradually declined once bleeding had been controlled (Table 1). Prothrombin time (PT) and INR were mildly elevated on admission, with a PT of 15.6 (12.1–15 seconds) and INR of 1.2. PT and INR rose when the patient decompensated (maximum 18.3 and 1.5, respectively) but subsequently returned to normal ranges by discharge. PTT was 34 (25–38 seconds) on admission and was never elevated. Factor X activity was also normal (95%, normal 81–157%).
Our patient was discharged medically stable, and treatment of AL amyloidosis was initiated. In AL amyloidosis, the efficacy of treatment is assessed by hematological response and organ response, both of which correlate with overall survival [4,5]. Hematological response is measured by trending the difference between the involved and uninvolved free light chain (dFLC), although negative serum and urine immunofixation for monoclonal protein is also required for a complete hematological response [4]. Our patient lacked significant light chain elevation, and at diagnosis the dFLC was already less than 40 mg/L (Table 2), which is the requirement to achieve a very good partial hematological response [4]. Therefore, we trended serum monoclonal protein as a marker of hematological response, in hopes of achieving the negative immunofixation required for a complete response [4]. Organ response, which is not currently graded and is classified only as present or absent, is determined by monitoring a marker of organ dysfunction that is specific to the organ known to be affected by amyloid disease [4]. For patients with hepatic AL amyloidosis, organ response is defined as a 50% decrease in an elevated alkaline phosphatase value or a decrease in liver size, measured radiographically, by at least 2 cm [4].
Our patient received 3 cycles of Cyclophosphamide-Bortezomib-Dexamethasone (CyBorD), resulting in a decline of serum monoclonal protein from a maximum of 0.99 g/dL to 0.47 g4L (Table 2) and a significant reduction in alkaline phosphatase from 923 to 327 (Figure 2). Although the decline in alkaline phosphatase achieved the >50% reduction needed for organ response, the patient had a persistent M spike and alkaline phosphatase was still elevated. Therefore, the anti-CD38 antibody daratumumab (DARA) was added to deepen the response. Our patient ultimately received a total of 8 cycles of CyBorD, 5 of which included DARA. After these 8 cycles, alkaline phosphatase declined to a nadir of 220 (Figure 2) and serum monoclonal protein declined to 0.29 g/dL (Table 2), indicating deepening of both hematological and organ response. The patient then underwent melphalan-based consolidation therapy with stem cell rescue, resulting in a further decrease of serum monoclonal protein to 0.18 g/dL and stable alkaline phosphatase (Table 2 and Figure 2, respectively). She is currently planned to undergo re-staging on post-transplant day 100, at which time maintenance treatment with lenalidomide will be considered.
Discussion
We report a case of AL amyloidosis with amyloid deposition confined to the liver and bone marrow, without significant light chain elevation, that responded to treatment with CyBorD plus daratumumab. Isolated hepatic involvement and lack of light chain elevation are both rare in AL amyloidosis. Approximately 10–20% of AL amyloidosis patients do not have significant serum free light chain production or an abnormal kappa/lambda chain ratio [6–8]. Likewise, our patient never had significant light chain secretion, and the difference between involved and uninvolved free light chains (dFLC) in our patient was consistently less than 40 mg/L (Table 2). Intriguingly, a dFLC of less than 50 mg/L is independently associated with improved survival, as well as a higher rate of complete hematological response [9]. The ideal method to monitor hematological response in patients with AL amyloidosis who have low light chain levels remains unclear. A “low dFLC partial response” has been defined as a decrease of dFLC to less than 10 in patients who present with a dFLC of 20–50 [9], and a low dFLC partial response correlates with overall survival [9]. However, as our patient consistently had a dFLC of less than 10, she could not be assessed for a low dFLC partial response. In the absence of guidelines, we opted to monitor hematological response by serum monoclonal protein.
In a large case series of 98 patients with hepatic AL amyloidosis, 33% of patients also presented with nephrotic syndrome, 12% also presented with congestive heart failure, and 11% also presented with peripheral neuropathy or carpal tunnel syndrome [10], suggesting that most patients with hepatic AL amyloidosis present with involvement of additional organ systems.
The diagnosis of hepatic AL amyloidosis, particularly hepatic amyloidosis without additional systemic findings, is challenging, with one study reporting that clinicians only considered the diagnosis of amyloid, before liver biopsy, in 26% of cases [10]. Among patients with hepatic amyloidosis, hepatomegaly and elevated alkaline phosphatase are the most common findings [10], but these are nonspecific, illustrating the difficulty of differentiating hepatic amyloidosis from more common etiologies of liver disease. Furthermore, a large retrospective study of 673 AL amyloidosis patients who were ultimately diagnosed by tissue biopsy showed that serum and urine immunofixation each only identified 78% of patients with disease [11]. Serum free light chains were also only abnormal in 77% of patients [11]. This suggests that when amyloidosis is suspected, a thorough laboratory investigation is required to rule out disease, and clinicians should not hesitate to perform bone marrow and tissue biopsy if clinical suspicion is high. Given that effective therapy for AL amyloidosis is available, delays in diagnosis represent a missed therapeutic opportunity to minimize end-organ damage and improve outcomes.
Treatment of AL amyloidosis consists of initial induction therapy with plasma cell targeting agents, after which myeloablative chemotherapy with stem cell rescue may be pursued [1]. Induction with CyBorD was selected for our patient due to the regimen’s high response rate, as clinical trials have shown initial hematological response rates as high as 94% [12]. Three cycles of CyBorD alone achieved the 50% reduction in alkaline phosphatase necessary for organ response (Figure 2) but did not result in the negative immunofixation necessary for complete hematological response [4]. Although organ response has been traditionally classified as either present or absent [4], the depth of organ response is associated with survival, and new graded organ response criteria are being considered [5]. Our patient therefore had the potential to deepen both hemato-logical and organ response with additional therapy.
Daratumumab, an anti-CD38 antibody, can induce hematological response and organ response in heavily pre-treated AL amyloidosis patients [13, 14]. Furthermore, the published “safety run-in” results from the ANDROMEDA study reported that the combination of CyBorD with daratumumab, in patients with newly diagnosed AL amyloidosis, produced a hematological response rate of 96%, a 36% complete hematological response rate, and an organ response rate of 64% [15]. Based on this evidence, we added daratumumab to CyBorD, and our patient received an additional 5 cycles of CyBorD+daratumumab (for a total of 8 treatment cycles). After induction with this regimen, alkaline phosphatase declined to a nadir of 220 U/L (Figure 2) and serum monoclonal protein declined to 0.29 g/dL (Table 2). By the graded organ response criteria, this decline in alkaline phosphatase constitutes a very good partial organ response (>60% reduction from baseline AP level to a nadir above ×2 lower limit of normal, with 2 times the lower limit of normal in our laboratory being equal to 76 U/L), suggesting that our patient benefited from the addition of daratumumab [5].
The benefits of autologous stem cell transplant in AL amyloidosis patients who have been treated with CyBorD and daratumumab remain poorly described. In the published safety and preliminary efficacy results from the ANDROMEDA study, 9 patients treated with CyBorD and daratumumab went on to receive stem cell transplant [15]. Among these patients, only 4 deepened their hematological response [15]. Our patient went on to receive melphalan-based consolidation therapy with stem cell rescue, which she tolerated well. Post-transplant serum monoclonal protein declined to 0.18 g/dL (Table 2) and alkaline phosphatase remained stable (Figure 2), suggesting a transplant-deepened hematological response. A related question is whether our patient will benefit from maintenance therapy after she reaches transplant day 100. The optimal timing of AL amyloidosis treatment remains unclear, with some advocating delaying treatment until the patient experiences organ progression or symptomatic relapse [16]. Moreover, a recent retrospective study suggests that lenalidomide or bortezomib maintenance post-transplant did not significantly improve overall survival or progression-free survival [17]. Ultimately, whether our patient pursues maintenance therapy will be informed by clinical judgement and shared decision making.
Conclusions
We describe a case of AL amyloidosis confined to the liver and bone marrow. This patient also lacked the light chain elevation that is considered a traditional hallmark of disease. Our patient responded to an induction regimen containing chemo-therapy and daratumumab, highlighting the effective treatment options for AL amyloidosis. Awareness that AL amyloidosis can have atypical presentations, including dysfunction in a single organ without certain expected laboratory findings, will help facilitate earlier diagnosis to optimize the efficacy of therapy.
Figures
Tables
Table 1.. Results obtained during admission confirming diagnosis of AL amyloidosis with amyloid deposition in the liver and bone marrow. All results were obtained prior to the initiation of treatment. Normal values, when applicable, are provided in the left-hand column. Table 2.. Markers of hematological response. Light chains were never significantly elevated, and as the difference between involved (lambda) and uninvolved (kappa) light chains was never greater than 40 mg/L, hematological response could not be assessed by tracking light chains. Our patient always had a normal kappa/lambda ratio. Therefore, we assessed the hematological response by trending serum monoclonal protein, which declined with CyBorD, the addition of DARA, and then reached a nadir after stem cell transplant.
Table 2.. Markers of hematological response. Light chains were never significantly elevated, and as the difference between involved (lambda) and uninvolved (kappa) light chains was never greater than 40 mg/L, hematological response could not be assessed by tracking light chains. Our patient always had a normal kappa/lambda ratio. Therefore, we assessed the hematological response by trending serum monoclonal protein, which declined with CyBorD, the addition of DARA, and then reached a nadir after stem cell transplant.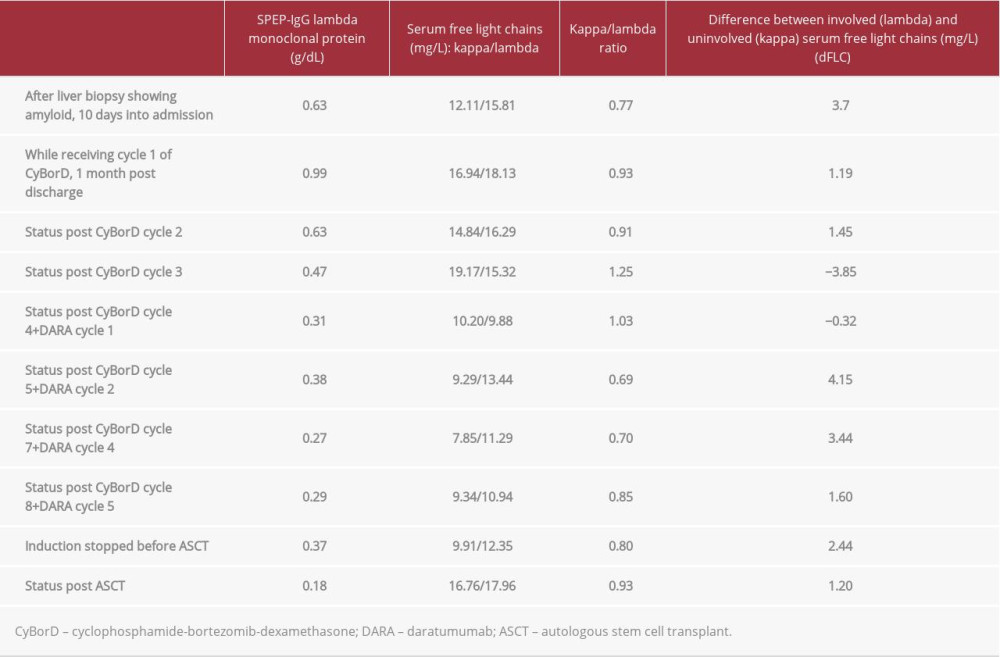
References:
1.. Gertz MA, Immunoglobulin light chain amyloidosis: 2018 Update on diagnosis, prognosis, and treatment: Am J Hematol, 2018; 93(9); 1169-80
2.. Rajkumar SV, Dimopoulos MA, Palumbo A, International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma: Lancet Oncol, 2014; 15(12); e538-48
3.. Merlini G, Seldin DC, Gertz MA, Amyloidosis: Pathogenesis and new therapeutic options: J Clin Oncol, 2011; 29(14); 1924-33
4.. Comenzo RL, Reece D, Palladini G, Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis: Leukemia, 2012; 26(11); 2317-25
5.. Muchtar E, Dispenzieri A, Leung N, Depth of organ response in AL amyloidosis is associated with improved survival: Grading the organ response criteria: Leukemia, 2018; 32(10); 2240-49
6.. Palladini G, Russo P, Bosoni T, Identification of amyloidogenic light chains requires the combination of serum-free light chain assay with immunofixation of serum and urine: Clin Chem, 2009; 55(3); 499-504
7.. Katzmann JA, Abraham RS, Dispenzieri A, Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice: Clin Chem, 2005; 51(5); 878-81
8.. Prokaeva T, Spencer B, Sun F, Immunoglobulin heavy light chain test quantifies clonal disease in patients with AL amyloidosis and normal serum free light chain ratio: Amyloid, 2016; 23(4); 214-20
9.. Dittrich T, Bochtler T, Kimmich C, AL amyloidosis patients with low amyloidogenic free light chain levels at first diagnosis have an excellent prognosis: Blood, 2017; 130(5); 632-42
10.. Park MA, Mueller PS, Kyle RA, Primary (AL) hepatic amyloidosis: Clinical features and natural history in 98 patients: Medicine (Baltimore), 2003; 82(5); 291-98
11.. Staron A, Kataria Y, Murray DL, Systemic AL amyloidosis with an undetectable plasma cell dyscrasia: A zebra without stripes: Am J Hematol, 2020; 95(2); E45-48
12.. Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Cyclophosphamidebortezomib-dexamethasone (CyBorD) produces rapid and complete hemato-logic response in patients with AL amyloidosis: Blood, 2012; 119(19); 4391-94
13.. Kaufman GP, Schrier SL, Lafayette RA, Daratumumab yields rapid and deep hematologic responses in patients with heavily pretreated AL amyloidosis: Blood, 2017; 130(7); 900-2
14.. Schwotzer R, Manz MG, Pederiva S, Daratumumab for relapsed or refractory AL amyloidosis with high plasma cell burden: Hematol Oncol, 2019; 37(5); 595-600
15.. Palladini G, Kastritis E, Maurer MS, Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: Safety run-in results of ANDROMEDA: Blood, 2020; 136(1); 71-80
16.. Sanchorawala V, Delay treatment of AL amyloidosis at relapse until symptomatic: Devil is in the details: Blood Adv, 2019; 3(2); 216-18
17.. Ozga M, Zhao Q, Benson D, AL Amyloidosis: The effect of maintenance therapy on autologous stem cell transplantation outcomes: J Clin Med, 2020; 9(11); 3778
Figures
Tables
Table 1.. Results obtained during admission confirming diagnosis of AL amyloidosis with amyloid deposition in the liver and bone marrow. All results were obtained prior to the initiation of treatment. Normal values, when applicable, are provided in the left-hand column.Table 2.. Markers of hematological response. Light chains were never significantly elevated, and as the difference between involved (lambda) and uninvolved (kappa) light chains was never greater than 40 mg/L, hematological response could not be assessed by tracking light chains. Our patient always had a normal kappa/lambda ratio. Therefore, we assessed the hematological response by trending serum monoclonal protein, which declined with CyBorD, the addition of DARA, and then reached a nadir after stem cell transplant.Table 1.. Results obtained during admission confirming diagnosis of AL amyloidosis with amyloid deposition in the liver and bone marrow. All results were obtained prior to the initiation of treatment. Normal values, when applicable, are provided in the left-hand column.Table 2.. Markers of hematological response. Light chains were never significantly elevated, and as the difference between involved (lambda) and uninvolved (kappa) light chains was never greater than 40 mg/L, hematological response could not be assessed by tracking light chains. Our patient always had a normal kappa/lambda ratio. Therefore, we assessed the hematological response by trending serum monoclonal protein, which declined with CyBorD, the addition of DARA, and then reached a nadir after stem cell transplant. In Press
06 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942937
12 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943244
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943275
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943411
Most Viewed Current Articles
07 Mar 2024 : Case report
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250